Activation et inhibition des voies de signalisation cellulaire

Table des Matières
Introduction
La signalisation cellulaire est le processus physiologique par lequel les cellules détectent et répondent aux signaux extracellulaires et intracellulaires via une séquence d'événements biochimiques comprenant la liaison ligand-récepteur, la transduction du signal par l'intermédiaire de seconds messagers et de réseaux protéiques, l'amplification, l'intégration et la terminaison pour produire des réponses cellulaires spécifiques telles que des changements dans l'expression des gènes, le métabolisme ou la motilité
Principe
Les voies de signalisation cellulaire peuvent être modulées par diverses approches chimiques, génétiques et biophysiques. Les modulateurs à petites molécules et les inhibiteurs de kinases activent ou inhibent sélectivement des cibles telles que les récepteurs ou les kinases. Les méthodes de chimio-génétique et de dimérisation permettent un contrôle rapide, réversible et inductible des complexes de signalisation. La surexpression ou l'inhibition génétique ajuste l'abondance des protéines pour amplifier ou supprimer l'activité des voies de signalisation. L'optogénétique utilise des domaines photosensibles pour contrôler la fonction des protéines avec une grande précision spatio-temporelle, tandis que la photoactivation et le photocage libèrent les molécules actives à la demande, permettant une régulation temporelle finement contrôlée de la signalisation.
Équipements, réactifs et consommables
|
ÉQUIPEMENT |
CONSOMMABLES |
|
|
|
|
|
RÉACTIFS Cellules et culture
Composés chimiques et modulateurs
Colorants fluorescents et réactifs d'analyse
Réactifs de génétique et de transfection
|
Facteurs de stimulation
Réactifs pour l'analyse des protéines Anticorps
|
Protocol
1. Modulateurs sélectifs à petites molécules
Mécanisme
Les modulateurs sélectifs à petites molécules exercent leurs effets sur les voies de signalisation en se liant directement à une cible définie (par exemple, un récepteur, une kinase ou une autre protéine de signalisation) et en modifiant son activité de façon contrôlée. Dans le cas des récepteurs couplés aux protéines G (RCPG) , les agonistes se lient au site orthostérique et stabilisent une conformation active du récepteur qui favorise le couplage à la protéine G et la production de seconds messagers en aval, tandis que les antagonistes bloquent cette activation et les agonistes inverses suppriment l'activité basale. Les modulateurs allostériques se lient à des sites spécifiques pour amplifier (modulateur allostérique positif) ou atténuer (modulateur allostérique négatif) la réponse du récepteur à son ligand endogène. Cette sélectivité résulte d' une complémentarité structurale avec le site de liaison et se traduit souvent par une modulation dose-dépendante des seconds messagers (par exemple, Ca²⁺, IP₁, AMPc) sans effets hors cible importants lorsqu'elle est bien caractérisée. La sélectivité fonctionnelle (« signalisation biaisée ») peut orienter un récepteur vers des voies en aval spécifiques (par exemple, protéine G vs. β-arrestine), permettant un réglage thérapeutique précis des résultats cellulaires.
Les principes moléculaires sous-jacents à l'activation, à l'antagonisme et à la modulation allostérique des RCPG, ainsi que leurs conséquences sur la signalisation Ca²⁺ en aval, sont illustrés schématiquement dans des modèles représentatifs de sélectivité fonctionnelle et de signalisation biaisée (Kenakin, Functional selectivity and biased receptor signaling , Nature Reviews Drug Discovery , 2011).
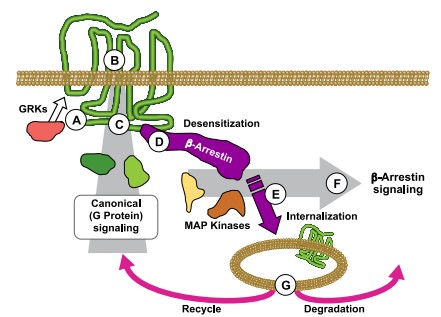
Figure 1 : La liaison d’un agoniste active les protéines G et induit la phosphorylation du récepteur par les GRK. Le récepteur agit comme une plateforme allostérique qui lie des protéines de signalisation intracellulaires et des modulateurs allostériques extracellulaires, modulant ainsi la signalisation en aval. Le recrutement de la β-arrestine favorise l’internalisation du récepteur et la formation de complexes de signalisation MAP kinase dans les endosomes, générant une signalisation cytosolique soutenue. Les récepteurs internalisés sont soit recyclés vers la membrane plasmique, soit dégradés, régulant ainsi la durée du signal.
Protocole expérimental : Test de mobilisation du Ca²⁺ pour les modulateurs des récepteurs couplés aux protéines G (RCPG)
Ce protocole suit un test standardisé de mobilisation du Ca²⁺ basé sur la fluorescence, utilisé pour identifier les agonistes, les antagonistes et les modulateurs allostériques des RCPG qui signalent via la libération intracellulaire de Ca²⁺.
Aperçu expérimental
Mesurer les variations de Ca²⁺ intracellulaire dans des cellules en culture exprimant un récepteur couplé aux protéines G (RCPG) d'intérêt en réponse à des composés testés. Les agonistes induisent une augmentation du Ca²⁺ cytosolique, les antagonistes bloquent les augmentations de Ca²⁺ induites par le ligand et les modulateurs allostériques modifient l'amplitude ou la cinétique de ces augmentations.
Étape 1 : Ensemencement et préparation des cellules
Les cellules exprimant le RCPG d'intérêt (par exemple, des cellules HEK293 exprimant de façon stable CXCR4 ou un autre récepteur couplé à la protéine Gq) sont ensemencées dans des plaques 96 ou 384 puits à fond transparent noir, à une densité optimisée pour atteindre une confluence de 70 à 90 % au moment du test (généralement 20 000 à 40 000 cellules/puits pour le format 96 puits). Les cellules sont incubées pendant une nuit (16 à 24 h) à 37 °C sous 5 % de CO₂ afin de permettre l'expression du récepteur et la récupération après le stress lié à l'ensemencement. Une confluence adéquate garantit une densité de récepteurs et une amplitude de signal homogènes entre les puits.
Étape 2 : Chargement du colorant (incorporation de l'indicateur Ca²⁺)
Le milieu de culture est retiré et remplacé par un tampon d'essai (par exemple, du HBSS supplémenté avec 20 mM d'HEPES, pH 7,4, et 2,5 mM de probénécide pour empêcher l'extrusion du colorant). Les cellules sont incubées avec un colorant fluorescent perméable à la membrane et sensible au Ca²⁺, tel que le Fluo-4 AM (concentration finale généralement de 2 à 5 µM), pendant 30 à 60 minutes à 37 °C. Durant cette incubation, le colorant diffuse dans les cellules et est clivé par des estérases intracellulaires en sa forme active, sensible au Ca²⁺.
Étape 3 : Désestérification et équilibration
Après le chargement du colorant, les cellules sont lavées une ou deux fois avec du tampon d'essai frais et incubées pendant 15 à 30 minutes supplémentaires à température ambiante ou à 37 °C afin de permettre la désestérification complète du colorant et la stabilisation de la fluorescence basale. Cette étape minimise la fluorescence de fond et améliore le rapport signal/bruit.
Étape 4 : Acquisition de la fluorescence de base
Les plaques sont placées dans un lecteur de plaques de fluorescence permettant des mesures cinétiques. La fluorescence basale est enregistrée pendant 30 à 120 secondes afin d'établir un signal de pré-stimulation stable (F₀). Cette valeur basale est essentielle à la normalisation et à la quantification précise des réponses calciques.
Étape 5 : Addition des composés et enregistrement cinétique
Les composés de test (agonistes, antagonistes ou modulateurs allostériques sélectifs à petites molécules) sont ajoutés rapidement à l'aide d'un injecteur automatisé tandis que l'acquisition de fluorescence se poursuit.
On ajoute les agonistes seuls pour évaluer l'activation directe de la voie de signalisation.
Les antagonistes sont pré-incubés (5 à 30 min) avant l'ajout d'un agoniste connu.
Les modulateurs allostériques sont co-appliqués ou pré-incubés selon leur mécanisme.
La fluorescence est enregistrée en continu pendant 2 à 5 minutes pour capturer la cinétique de libération et de décroissance du pic de Ca²⁺.
Mesures de contrôle de la qualité
Contrôles de performance des essais
- Agoniste témoin positif : un ligand connu du récepteur confirme le couplage fonctionnel du récepteur à la signalisation Ca²⁺. L’absence de réponse indique une expression anormale du récepteur ou un échec du test.
- Contrôle négatif (véhicule) : définit la variabilité de fluorescence de base et exclut les effets du solvant (par exemple, DMSO ≤ 0,1 %).
Interprétation des résultats
Activation induite par un agoniste
Une augmentation rapide et transitoire de la fluorescence (ΔF/F₀) indique la libération de Ca²⁺ à partir des réserves intracellulaires suite à l'activation du RCPG. Les courbes dose-réponse sont ajustées pour déterminer les valeurs EC₅₀, reflétant la puissance de l'agoniste, tandis que la réponse maximale (Emax) reflète son efficacité.
Inhibition médiée par un antagoniste
Les antagonistes réduisent ou suppriment le signal Ca²⁺ induit par l'agoniste. Les antagonistes compétitifs provoquent un déplacement vers la droite de la courbe dose-réponse de l'agoniste sans réduire l'Emax, tandis que les antagonistes non compétitifs réduisent l'Emax. Les valeurs IC₅₀ quantifient la force de l'inhibition.
Modulation allostérique
Les modulateurs allostériques modifient les réponses aux agonistes sans activer directement le récepteur. Les modulateurs allostériques positifs augmentent l'amplitude ou la puissance du signal (déplacement vers la gauche), tandis que les modulateurs négatifs atténuent les réponses. Les modifications de la pente de la courbe ou de la réponse maximale sont caractéristiques des mécanismes allostériques.
2. Sondes chimiques et inhibiteurs de kinases
Mécanisme
Les sondes chimiques sont de petites molécules puissantes et sélectives conçues pour moduler l'activité d'une cible spécifique au sein d'un système biologique, comme une kinase particulière, et servent d'outils pour élucider les mécanismes biologiques. Les inhibiteurs de kinase constituent une sous-classe de ces sondes ; ils bloquent l'activité catalytique de la kinase en se liant à des sites fonctionnels critiques, tels que le site de liaison de l'ATP, afin d'empêcher la phosphorylation. Ils comprennent les inhibiteurs compétitifs de l'ATP qui occupent le site de liaison de l'ATP, les inhibiteurs allostériques ciblant des sites situés en dehors du site actif, les inhibiteurs covalents formant des liaisons irréversibles avec la kinase, et les dégradeurs induisant la dégradation de la protéine cible.
Protocole expérimental : Inhibition de MEK avec l’U0126 (un inhibiteur de kinase)
Aperçu expérimental
La voie MAPK/ERK transmet les signaux mitogènes de la membrane cellulaire au noyau par une succession d'étapes de phosphorylation. L'U0126 est un inhibiteur chimique qui bloque sélectivement l'activité de la kinase MEK1/2, empêchant ainsi la phosphorylation d'ERK et les effets de signalisation en aval.
Voici un protocole expérimental détaillé, étape par étape, sur des cellules en culture pour mesurer l'inhibition de la voie MAPK/ERK par l'inhibiteur de MEK U0126 afin d'évaluer comment U0126 inhibe l'activité de MEK1/2 et réduit par conséquent la phosphorylation d'ERK1/2 dans les cellules en culture en utilisant Western blot pour quantifier les niveaux de phospho-ERK (p-ERK).
Étape 1 : Préparation des cellules
Ensemencer les cellules dans des plaques à 6 puits à une confluence d'environ 60 à 70 % et incuber pendant la nuit pour qu'elles adhèrent.
Étape 2 : Privation de sérum
Remplacer le milieu de culture par un milieu sans sérum pendant 12 à 16 heures afin de réduire la signalisation basale et de synchroniser les cellules.
(Optionnel : permet une meilleure détection de l’induction du signal et des effets des inhibiteurs.)
Étape 3 : Traitement inhibiteur
- Préparer des dilutions de U0126 (par exemple, 0,1, 1, 5, 10 µM) dans un milieu de culture.
- Ajouter l'inhibiteur aux puits ; inclure un témoin véhicule (équivalent DMSO).
- Incuber les cellules avec l'inhibiteur pendant 15 à 30 min à 37 °C. Cela permet à l'U0126 de se lier à MEK et d'inhiber son activité.
Étape 4 : Stimulation optionnelle
Pour activer la signalisation, il est possible d'ajouter un facteur de croissance (par exemple, l'EGF) pendant 5 à 15 minutes.
Cela augmente l'activation de la voie de signalisation et offre une plage dynamique pour évaluer l'inhibition.
Étape 5 : Lyse cellulaire
- Laver les cellules avec du PBS froid.
- Lyser les cellules sur glace avec un tampon de lyse + des inhibiteurs pour préserver la phosphorylation.
Étape 6 : Western Blot
- Mesurer la concentration en protéines (par exemple, dosage BCA), charger des quantités égales (20–40 µg).
- Effectuer une électrophorèse SDS-PAGE et transférer sur PVDF.
- Sonder avec un anticorps anti-phospho-ERK1/2, puis normaliser avec l'ERK1/2 total et le contrôle de charge.
- Détecter les bandes par chimiluminescence et imagerie.
Le protocole complet de Western blot étape par étape est décrit en détail dans ce protocole .
Quantification:
Utiliser la densitométrie pour mesurer l'intensité des bandes p-ERK normalisée par rapport à l'ERK totale.
Les comparaisons quantitatives entre les différentes doses d'inhibiteur indiquent l'efficacité de l'inhibition.
Mesures de contrôle de la qualité
Un contrôle qualité rigoureux est essentiel pour interpréter les expériences d'inhibition de la kinase.
-
Contrôle du véhicule (DMSO uniquement) : Détermine la phosphorylation de base en l'absence d'inhibiteur.
-
Aucun contrôle de stimulation (si un facteur de croissance est utilisé) : établit l’activation basale d’ERK.
-
Contrôle d'activation positive : les cellules traitées avec un facteur de croissance sans inhibiteur doivent présenter une élévation de p-ERK.
-
Concentrations multiples d'inhibiteurs : confirme la relation dose-réponse.
L'inhibition dose-dépendante de la phosphorylation intracellulaire d'ERK par U0126 dans les cellules COS-7 a été démontrée dans l'identification d'un nouvel inhibiteur de la kinase de protéine activée par les mitogènes (Favata et al. , Journal of Biological Chemistry, 1998).
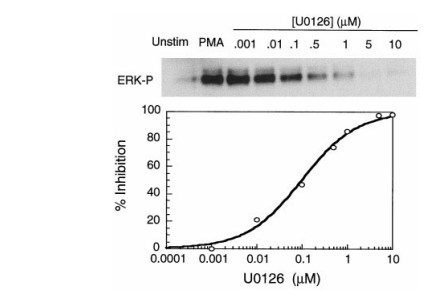
Fig2 : Dans les cellules COS-7 stimulées par le phorbol 12-myristate 13-acétate (PMA), la phosphorylation d'ERK1/2 a été fortement induite, tandis que le prétraitement avec l'inhibiteur de MEK U0126 a entraîné une suppression marquée et dose-dépendante des niveaux de phospho-ERK, cohérente avec l'inhibition sélective de l'activité de MEK .
Interprétation des résultats
Modèle de résultats attendus
- Véhicule + facteur de croissance : Niveaux élevés de p-ERK dus à l’activation de la voie.
- Traitement U0126 : Diminution dose-dépendante de la p-ERK , avec un effet minimal sur l’ERK totale.
Cela indique une inhibition efficace de MEK empêchant la signalisation en aval.
Si le p-ERK ne diminue pas, interprétations possibles :
- Concentration d'inhibiteur trop faible.
- Les effets hors cible sont prédominants.
- Activation de la voie indépendante de MEK.
3. Méthodes de génétique chimique et de dimérisation
Mécanisme
Les méthodes de génétique chimique et de dimérisation induite sont des approches de biologie chimique qui permettent une activation ou une inhibition aiguë, réversible et spatialement contrôlée des voies de signalisation cellulaire grâce à des interactions protéine-petite molécule conçues à cet effet, plutôt qu'à la liaison de ligands endogènes ou à l'inhibition enzymatique.
La génétique chimique repose sur la modification d'une protéine de signalisation (généralement une kinase ou un effecteur de signalisation) pour la rendre particulièrement sensible à une petite molécule synthétique qui n'affecte pas le protéome de type sauvage.
La dimérisation chimiquement induite (CID) utilise des petites molécules bifonctionnelles ou monovalentes (par exemple, la rapamycine) pour induire la proximité entre des domaines protéiques modifiés (par exemple, FKBP et FRB). La dimérisation forcée peut activer la signalisation en recrutant des enzymes sur des substrats ou des membranes , ou l'inhiber par séquestration ou mauvaise localisation . La CID permet un contrôle temporel de l'ordre de la seconde à la minute et peut être inversée par lavage ou par des ligands compétitifs.
Protocole expérimental : Activation par dimérisation induite chimiquement de la voie PI3K–Akt
La voie PI3K–Akt est une cascade de signalisation essentielle à la survie et à la croissance, activée au niveau de la membrane plasmique. L'activation d'Akt nécessite le recrutement de PI3K à la membrane, la production de PIP3 et la phosphorylation subséquente d'Akt.
Dans ce protocole, l'hétérodimérisation induite par la rapamycine est utilisée pour activer artificiellement la voie de signalisation PI3K en recrutant la sous-unité catalytique PI3K (p110) à la membrane plasmique via l'interaction FKBP–FRB, indépendamment de la stimulation du récepteur. L'activation d'Akt est évaluée par la mesure de l'Akt phosphorylée (Ser473) par Western blot.
Cette stratégie a été initialement démontrée pour établir la suffisance de la localisation membranaire de PI3K pour l'activation d'Akt .
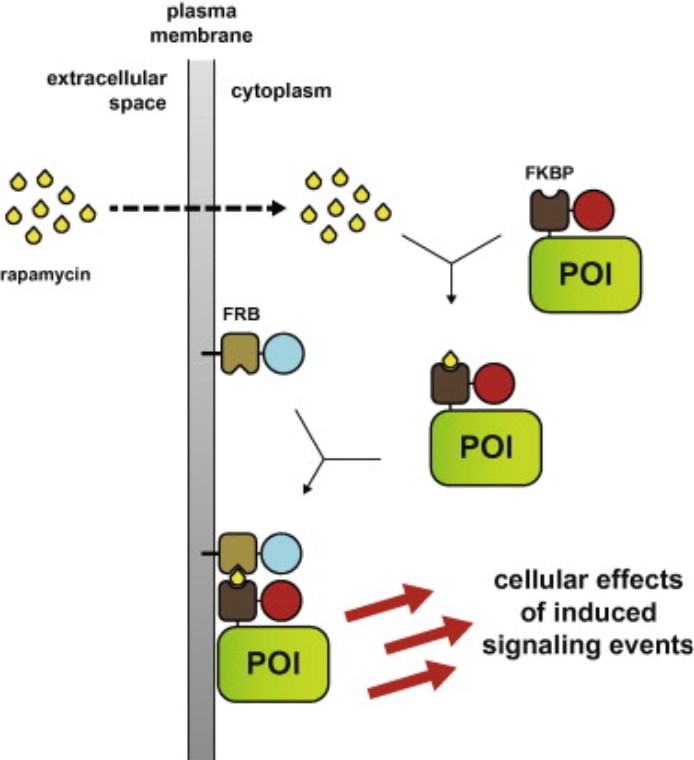
Figure 3 : La rapamycine induit l'hétérodimérisation FKBP–FRB, recrutant des protéines de signalisation marquées FKBP sur les membranes ancrées à FRB, ce qui conduit à une activation rapide des voies en aval telles que la phosphorylation d'Akt. (Putyrski, M., & Schultz, C. Protein translocation as a tool: The current rapamycin story . FEBS Letters, 2012).
Étape 1 : Préparation cellulaire et expression des composants du CID
-
Cultiver des cellules de mammifères (par exemple, HEK293 ou NIH-3T3) dans des conditions standard (37 °C, 5 % CO₂).
-
Transfecter les cellules avec les constructions suivantes :
-
FRB-CAAX (FRB fusionné à un motif de ciblage de la membrane plasmique)
-
FKBP-p110 (FKBP fusionné à la sous-unité catalytique de PI3K)
-
-
Incuber les cellules pendant 24 à 48 h pour permettre une expression protéique suffisante.
Étape 2 : Privation de sérum
-
Remplacer le milieu de culture par un milieu sans sérum.
-
Incuber pendant 12 à 16 h.
pour réduire l'activité basale de PI3K–Akt et améliorer le rapport signal/bruit pour l'activation chimique.
Étape 3 : Dimérisation induite chimiquement
-
Préparer une solution de travail de rapamycine (concentration finale : 10–100 nM).
-
Ajouter la rapamycine directement au milieu de culture.
-
Incuber pendant 5 à 30 min à 37 °C.
La rapamycine se lie simultanément à FKBP et FRB, induisant une hétérodimérisation et recrutant PI3K à la membrane plasmique , initiant ainsi la production de PIP3.
Étape 4 : Lyse cellulaire
-
Laver rapidement les cellules avec du PBS glacé.
-
Lyser les cellules sur glace à l'aide d'un tampon de lyse RIPA ou NP-40 supplémenté avec :
-
Inhibiteurs de protéase
-
Inhibiteurs de phosphatase
-
-
Clarifier les lysats par centrifugation.
Étape 5 : Analyse par Western Blot
-
Quantifier la concentration en protéines (par exemple, dosage BCA).
-
Charger des quantités égales de protéines (20–40 µg) par voie.
-
Effectuer une électrophorèse SDS-PAGE et transférer sur une membrane PVDF.
-
Sonder avec :
-
Anti-phospho-Akt (Ser473)
-
Anti-total Akt
-
Contrôle de charge (β-actine ou GAPDH)
-
-
Détecter les signaux par chimiluminescence.
Étape 6 : Quantification
-
Effectuer une analyse densitométrique.
-
Normaliser le phospho-Akt par rapport à l'Akt total.
-
Comparer les échantillons traités à la rapamycine aux échantillons non traités.
Mesures de contrôle de la qualité
Un contrôle qualité rigoureux est essentiel pour interpréter les expériences de signalisation basées sur le CID.
- Contrôle sans rapamycine :
Confirme que l'activation de la signalisation est strictement induite chimiquement.
- Commandes à construction unique :
Les cellules exprimant uniquement FKBP-p110 ou FRB-CAAX ne devraient pas activer Akt.
- Titrage de la dose de rapamycine :
Garantit que l'activation dépend de la concentration.
- Analyse de l'évolution temporelle :
Présente une cinétique rapide compatible avec une activation directe de la voie.
- Contrôles de chargement et d'expression :
Vérifier l'égalité de la charge protéique et l'expression comparable des composants CID.
Interprétation des résultats
Modèle de résultats attendus
- Cellules non traitées :
Faible phosphorylation basale d’Akt due à l’absence de recrutement membranaire de PI3K. - Cellules traitées à la rapamycine :
augmentation rapide et robuste de la phosphorylation d’Akt Ser473 sans stimulation par facteur de croissance.
Ceci démontre que la localisation forcée de PI3K est suffisante pour activer la voie PI3K–Akt , confirmant la causalité entre l'organisation spatiale et la sortie du signal.
4. Outils optogénétiques et à codage génétique
Mécanisme
Dans cette étude, un système optogénétique a été conçu pour contrôler le module Ras/Erk en couplant des modules d'hétérodimérisation photo-induits à un domaine GEF (facteur d'échange de nucléotides guanine) de Ras. Cette approche utilise le système de dimérisation photo-inductible Phytochrome B (PhyB)/PIF (facteur d'interaction avec le phytochrome) : une molécule de PhyB membranaire fusionnée à un motif CAAX et une molécule de PIF cytosolique fusionnée au domaine catalytique de l'activateur de Ras, SOS (SOScat). La lumière rouge induit l'hétérodimérisation de PhyB et PIF, recrutant SOScat à la membrane plasmique où il active Ras endogène, initiant ainsi la cascade de kinases RAF–MEK–ERK. Lorsque la lumière activatrice est éteinte ou alternée avec des conditions inactivatrices, la dimérisation s'inverse, inhibant l'activation de Ras et permettant la diminution de la phosphorylation d'ERK. Cette stratégie isole le module Ras/Erk des récepteurs en amont et des ligands naturels afin d'étudier sa dynamique de transmission intrinsèque.
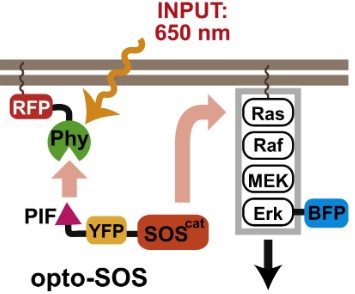
Figure 4 : La lumière rouge recrute PIF-SOScat à la protéine PhyB membranaire, activant Ras et induisant la translocation nucléaire de BFP-Erk2. (Toettcher, JE, et al. Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 2013)
Protocole expérimental : Activation optogénétique du module Ras/Erk
Aperçu expérimental
L'objectif est d'utiliser la lumière rouge pour recruter opto-SOS (PIF-SOScat) à la membrane plasmique via PhyB–CAAX, activant ainsi Ras et permettant de suivre la dynamique de phosphorylation d'Erk dans des cellules en culture. Les critères d'évaluation comprennent la détection par Western blot de la phospho-Erk et l'imagerie de cellules vivantes avec un rapporteur d'activité d'Erk.
Étape 1 : Préparation des cellules
Ensemencer une lignée cellulaire de mammifères (par exemple, des cellules NIH 3T3 ou PC12) dans des plaques à six puits ou des boîtes de Petri à fond de verre à une confluence d'environ 60 à 70 %, et incuber pendant une nuit à 37 °C avec 5 % de CO₂. Une confluence optimale garantit une transfection efficace et une exposition uniforme à la lumière.
Étape 2 : Transfection des constructions optogénétiques
Transfecter les cellules avec deux constructions :
-
Une fusion PhyB-mCherry-CAAX localisée dans la membrane plasmique , qui ancre le récepteur de lumière rouge à la membrane.
-
Une fusion PIF-SOScat-YFP , dans laquelle le domaine PIF est fusionné au domaine catalytique du GEF Ras SOS et marqué avec YFP.
Utiliser un réactif de transfection à base de lipides et incuber les cellules pendant 24 à 48 heures pour permettre l'expression.
Justification : Le domaine SOScat est dépourvu de ciblage membranaire intrinsèque ; son recrutement à la membrane par PhyB en réponse à la lumière rouge permet une activation spécifique de Ras indépendante de la stimulation du récepteur.
Étape 3 : Mesure de référence et privation de sérum
Remplacer le milieu de culture par un milieu sans sérum pendant 12 à 16 heures avant l'illumination afin de réduire l'activité basale de Ras/Erk, améliorant ainsi la détection des changements de signalisation induits optogénétiquement.
Étape 4 : Stimulation lumineuse optogénétique
Placer les cellules sous un système d'éclairage LED programmable capable de délivrer des impulsions de lumière rouge de 650 nm , qui déclenchent l'hétérodimérisation PhyB–PIF. Éclairer avec des impulsions de lumière rouge (650 nm) de 1 à 2 minutes, suivies de lumière rouge lointaine (750 nm) pour inverser la liaison si nécessaire.
-
Lumière rouge (650 nm) : recrute PIF-SOScat à la membrane → activation de Ras.
-
Lumière rouge lointaine (750 nm) : inverse l'association PhyB–PIF → SOScat retourne dans le cytosol.
Les protocoles d'illumination peuvent être modulés en termes de durée et de fréquence des impulsions afin d'étudier la dynamique. La liaison PhyB–PIF induit le recrutement de SOScat à la membrane, activant Ras et déclenchant la phosphorylation d'Erk en aval.
Étape 5 : Échantillonnage temporel
Après des régimes d'illumination définis, collecter des cellules à plusieurs moments (par exemple, 0 min, 2 min, 5 min, 15 min, 30 min) pour capturer la dynamique de transmission du signal.
Étape 6 : Lyse cellulaire
Laver rapidement les cellules avec du PBS froid et les lyser sur glace à l'aide d'un tampon de lyse contenant des inhibiteurs de protéases et de phosphatases. Clarifier les lysats par centrifugation.
Étape 7 : Détection de l'activation de la signalisation
Mesure de l'activation d'Erk par Western blot :
-
Quantifier la concentration protéique par la méthode BCA ou un dosage similaire.
-
Déposer des quantités égales (20–40 µg) sur SDS-PAGE et transférer sur une membrane PVDF.
-
Sonder avec un anticorps anti-phospho-Erk1/2 puis anti-Erk1/2 total pour normalisation.
-
Détection par chimiluminescence.
Il est possible, en option, d'imager des cellules vivantes exprimant un rapporteur d'activité Erk fluorescent pour suivre la dynamique de localisation.
Mesures de contrôle de la qualité
-
Valider l'expression et la localisation des constructions optogénétiques par imagerie de fluorescence : PhyB-mCherry-CAAX doit se localiser au niveau de la membrane plasmique et PIF-SOScat-YFP doit présenter une distribution cytosolique à l'obscurité. Une expression insuffisante ou une mauvaise localisation compromet la réponse à la lumière.
-
Inclure des témoins sans stimulation lumineuse pour mesurer l'activité basale d'Erk. Inclure des cellules transfectées avec un seul vecteur pour confirmer que les composants PhyB et PIF sont tous deux nécessaires à la signalisation dépendante de la lumière.
-
S'assurer que les niveaux totaux d'Erk restent constants dans toutes les conditions afin de valider que les changements de phospho-Erk sont dus à l'activation de la voie et non à une abondance protéique altérée.
-
Effectuer plusieurs réplicats biologiques (≥3) pour assurer la reproductibilité et évaluer la variabilité des réponses de signalisation.
Interprétation des résultats
Comportement attendu : Sous illumination par lumière rouge, le recrutement de PIF-SOScat à la membrane devrait activer Ras et induire une phosphorylation accrue d’Erk1/2 par rapport aux témoins à l’obscurité. Les niveaux de phospho-Erk sont corrélés à la durée et à l’intensité de l’illumination. Le passage à une lumière rouge lointaine devrait inverser l’association PhyB-PIF, entraînant la dissociation de SOScat de la membrane et une diminution progressive des niveaux de phospho-Erk.
Si la stimulation optogénétique n'augmente pas la phosphorylation d'Erk, cela peut s'expliquer par une expression insuffisante des constructions, une intensité lumineuse inadéquate ou un timing incorrect. Si le niveau de phospho-Erk reste élevé après l'arrêt de la stimulation lumineuse, cela peut indiquer une cinétique de dissociation lente ou une rétroaction de signalisation secondaire.
5. Surexpression/inhibition génétique
Mécanisme
La surexpression et l'inhibition génétiques modulent la voie JAK/STAT en modifiant l'abondance des protéines de signalisation. La surexpression introduit de l'ADN exogène codant pour des composants clés, tels que STAT3, JAK1 ou des mutants constitutifs actifs, ce qui entraîne une augmentation de l'activité de la voie. L'inhibition utilise des siRNA ou des shRNA pour réduire l'expression de ces protéines, inhibant ainsi la transmission du signal.
Dans la voie JAK/STAT, les cytokines telles que l'IL-6 ou l'IFN-γ se lient à leurs récepteurs, activant ainsi les kinases JAK associées à ces récepteurs. Les JAK phosphorylent les facteurs de transcription STAT, qui se dimérisent et migrent vers le noyau pour réguler l'expression des gènes. La surexpression de STAT3 peut induire une translocation nucléaire et une transcription accrues, même en l'absence de stimulation par un ligand, tandis que l'inhibition de STAT3 ou de JAK1 réduit les réponses transcriptionnelles induites par les cytokines. Ces approches permettent une évaluation directe de la fonction de chaque composant de la voie.
Protocole expérimental : Inhibition génétique de STAT3 pour bloquer la voie de signalisation JAK/STAT
Aperçu expérimental
La voie JAK/STAT transmet les signaux des cytokines pour réguler les réponses immunitaires, la prolifération et la survie cellulaires. Dans ce protocole, l'inhibition de l'expression de STAT3 par siRNA est utilisée pour inhiber sa phosphorylation et sa translocation nucléaire. L'activité de la voie est mesurée par Western blot de phospho-STAT3 (Tyr705) et, optionnellement, par un test de luciférase rapporteur sensible à STAT.
Étape 1 : Préparation des cellules
Des lignées cellulaires humaines telles que HeLa, HepG2 ou U266 sont ensemencées dans des plaques à 6 puits à une confluence de 50 à 60 % et incubées pendant une nuit à 37 °C sous une atmosphère à 5 % de CO₂. Une confluence cellulaire adéquate garantit une efficacité de transfection optimale et une croissance cellulaire saine.
Étape 2 : Préparation des siRNA
Préparer des molécules d'ARNsi ciblant STAT3 à une concentration finale de 10 à 50 nM dans un milieu sans sérum. Inclure les contrôles suivants :
- Un siRNA non ciblé et brouillé pour contrôler les effets non spécifiques de la transfection
- Un siRNA de contrôle positif optionnel ciblant JAK1 pour vérifier la dépendance à la voie de signalisation
L'ARNsi induira la dégradation de l'ARNm de STAT3 par le complexe RISC, entraînant une réduction de l'expression protéique.
Étape 3 : Transfection d’ARNsi
Mélanger l'ARNsi avec un réactif de transfection lipidique et incuber brièvement pour permettre la formation de complexes de transfection. Ajouter ces complexes goutte à goutte aux cellules et incuber pendant 48 à 72 heures pour permettre une inhibition efficace de la protéine STAT3.
Étape 4 : Privation de sérum
Remplacer le milieu de culture par un milieu sans sérum et incuber les cellules pendant 12 à 16 heures afin de supprimer la phosphorylation basale de STAT3. Cette étape de synchronisation augmente la plage dynamique de détection des effets de l'inhibition.
Étape 5 : Stimulation par les cytokines
Stimuler les cellules avec des cytokines telles que l'IL-6 (10–20 ng/mL) ou l'IFN-γ pendant 15–30 minutes à 37 °C afin d'activer STAT3 dans les cellules témoins. Cette étape permet une comparaison directe entre les conditions d'inhibition et les conditions témoins.
Étape 6 : Lyse cellulaire
Laver délicatement les cellules avec du PBS glacé pour éliminer le milieu de culture. Lyser les cellules sur glace à l'aide d'un tampon de lyse additionné d'inhibiteurs de protéases et de phosphatases afin de préserver l'intégrité des protéines et leur état de phosphorylation. Clarifier les lysats par centrifugation et recueillir les surnageants pour analyse.
Étape 7 : Analyse par Western Blot
Déterminer la concentration protéique et déposer 20 à 40 µg par puits pour l'électrophorèse SDS-PAGE. Transférer les protéines séparées sur une membrane PVDF et incuber successivement avec les anticorps suivants :
- Anticorps anti-phospho-STAT3 (Tyr705) pour détecter la STAT3 activée
- Anticorps anti-STAT3 total pour la normalisation
- Anti-β-actine ou GAPDH comme témoin de charge
Détecter les bandes par chimiluminescence et enregistrer les images.
Étape 8 : Quantification des résultats
Effectuer une analyse densitométrique de l'intensité de la bande phospho-STAT3 normalisée par rapport à la STAT3 totale. Comparer les conditions d'inhibition à des contrôles siRNA non spécifiques sur l'ensemble des réplicats biologiques afin d'évaluer l'inhibition de la voie de signalisation.
Contrôle de la qualité et contrôles expérimentaux
-
Validation de l'inhibition : confirmer la réduction de la protéine STAT3 par Western blot ou qRT-PCR. Une inhibition incomplète peut fausser l'interprétation des résultats.
-
Contrôle négatif : un siRNA non spécifique établit l’activité de base de la voie de signalisation.
-
Contrôle d'activation positive : les cellules traitées avec une cytokine sans inhibition doivent présenter un signal phospho-STAT3 robuste.
-
Contrôle de chargement : s'assurer que les niveaux totaux de STAT3 ou de β-actine restent inchangés pour confirmer un chargement égal.
-
Réplicats : Effectuer au moins trois réplicats biologiques indépendants pour garantir la reproductibilité
Interprétation des résultats
Profil des résultats attendus
- Les cellules traitées avec un siRNA non spécifique et une stimulation par cytokines présentent des niveaux élevés de phospho-STAT3.
- Les cellules présentant une inhibition de STAT3 et une stimulation par cytokines montrent une réduction significative de la phospho-STAT3, tandis que la STAT3 totale reste inchangée.
Interprétation : L'inactivation efficace démontre que STAT3 est essentiel à la signalisation JAK/STAT et à la transcription induite par les cytokines.
Résultats alternatifs :
- L'absence de réduction de la phospho-STAT3 peut indiquer une inhibition incomplète ou une signalisation compensatoire par d'autres membres de la famille STAT.
- Une diminution du taux total de STAT3 indique des effets hors cible ou une toxicité cellulaire et invalide les conclusions.
6. Photoactivation
Mécanisme
La photoactivation est une approche physico-chimique qui utilise la lumière pour déclencher une réaction photochimique transformant une molécule inerte en une molécule active de signalisation . Dans les études de signalisation cellulaire, on utilise généralement des composés photolabiles , c'est-à-dire des molécules bioactives rendues inactives par un groupe protecteur photolabile . Sous l'effet d'une illumination à une longueur d'onde spécifique, le groupe protecteur est clivé, libérant ainsi la molécule active avec un contrôle temporel et spatial précis.
Contrairement à l'optogénétique, la photoactivation ne repose pas sur des protéines photosensibles codées génétiquement . Elle manipule directement les voies de signalisation endogènes en libérant des seconds messagers, des ligands ou des inhibiteurs natifs , tels que les ions calcium, les nucléotides cycliques, l'ATP ou les neurotransmetteurs.
Dans la signalisation calcique, les chélateurs de Ca²⁺ photolabiles, tels que le NP-EGTA ou le DM-nitrophène, fixent fortement le calcium et l'empêchent d'activer les effecteurs en aval. La lumière UV ou proche UV clive le groupe photolabile, libérant rapidement le Ca²⁺. L'augmentation soudaine du calcium intracellulaire active des protéines de signalisation calcium-dépendantes, comme la calmoduline et la CaMKII , induisant la phosphorylation de cibles en aval.
Étant donné que le démasquage est généralement irréversible , la photoactivation constitue une méthode puissante pour initier la signalisation avec une grande précision temporelle, mais une réversibilité limitée.
Protocole expérimental : Photoactivation de la signalisation Ca²⁺/CaMKII à l'aide de calcium encapsulé
Aperçu expérimental
La voie de signalisation Ca²⁺ /CaMKII régule la plasticité synaptique, la contraction musculaire et les réponses transcriptionnelles. L'activation de CaMKII nécessite une augmentation rapide et localisée du calcium intracellulaire.
Dans ce protocole, les cellules sont chargées avec du NP-EGTA-AM , un chélateur de calcium encapsulé perméable à la membrane. Sous illumination UV, le Ca²⁺ est libéré à l'intérieur de la cellule, activant la CaMKII. L'activation de la voie est quantifiée par détection de la phospho-CaMKII (Thr286) par Western blot .
Étape 1 : Préparation des cellules
Ensemencer des cellules en culture (HEK293, neurones ou cardiomyocytes) dans des plaques 6 puits ou des boîtes de Petri à fond de verre jusqu'à une confluence d'environ 60 à 70 % . Incuber pendant une nuit à 37 °C sous 5 % de CO₂ pour permettre l'adhésion et la récupération.
Étape 2 : Chargement des cellules avec du calcium encapsulé
Préparer le NP-EGTA-AM à une concentration finale de 2 à 10 µM dans un milieu sans sérum contenant du Pluronic F-127 pour améliorer la perméabilité membranaire.
Incuber les cellules avec le composé encapsulé pendant 30 à 45 minutes à 37 °C. Pendant ce temps, le NP-EGTA-AM diffuse dans les cellules et est clivé par les estérases intracellulaires, piégeant ainsi le chélateur encapsulé à l'intérieur de la cellule.
Étape 3 : Rinçage et équilibration
Laver délicatement les cellules avec une solution saline physiologique tamponnée pour éliminer le NP-EGTA-AM extracellulaire. Incuber les cellules pendant 15 à 30 minutes supplémentaires pour permettre une désestérification et un équilibrage complets.
Cette étape garantit que la libération de calcium lors de l'illumination provient des réserves intracellulaires.
Étape 4 : Photoactivation par exposition à la lumière
Exposer les cellules à la lumière UV (généralement 350–365 nm) à l'aide d'une source lumineuse montée sur un microscope ou d'une LED UV pendant une durée définie, généralement de quelques millisecondes à quelques secondes en fonction de l'amplitude de calcium souhaitée.
L'illumination clive le groupe photolabile du NP-EGTA, libérant ainsi du Ca²⁺ dans le cytosol. Cette augmentation soudaine de calcium active la calmoduline, qui à son tour active la CaMKII par autophosphorylation au niveau de la Thr286.
Étape 5 : Échantillonnage temporel
Après photoactivation, prélever des cellules à des moments précis, tels que 0, 30 secondes, 2 minutes, 5 minutes et 15 minutes , afin de capturer la dynamique de signalisation.
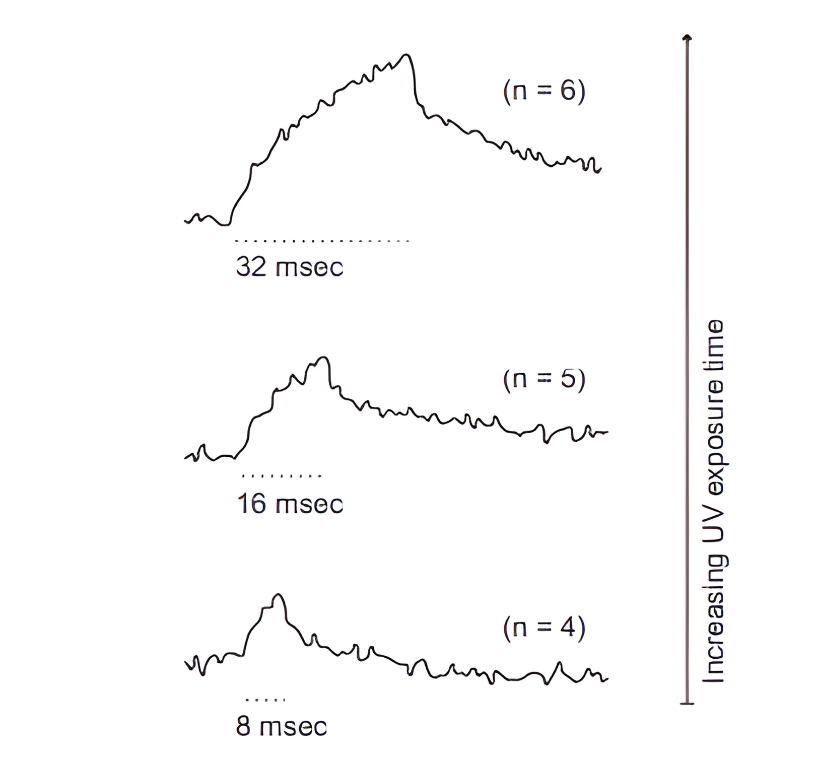
Figure 6 : Cette figure montre que la libération de NP-EGTA par UV induit des variations transitoires de Ca²⁺ intracellulaire en fonction du temps, les durées d’exposition plus longues produisant des augmentations de calcium progressivement plus importantes, démontrant ainsi un contrôle quantitatif de la libération de Ca²⁺ par la durée de libération. (Eder, A., & Bading, H. Calcium signals can freedom cross the nuclear envelope in hippocampal neurons: somatic calcium increases generate nuclear calcium transients. BMC Neuroscience, 2007).
Étape 6 : Lyse cellulaire
Laver rapidement les cellules avec du PBS glacé et les lyser sur glace en utilisant un tampon de lyse à base de détergent supplémenté en inhibiteurs de protéases et de phosphatases pour préserver les états de phosphorylation.
Clarifier les lysats par centrifugation et recueillir les surnageants pour analyse.
Étape 7 : Analyse par Western Blot
Mesurer la concentration protéique et déposer 20 à 40 µg par puits sur les gels SDS-PAGE. Transférer les protéines sur des membranes PVDF et les sonder avec :
-
Anticorps anti-phospho-CaMKII (Thr286)
-
Anticorps anti-CaMKII total
-
Contrôle de chargement tel que la β-actine
Détection des signaux par chimiluminescence et imagerie.
Quantification
Effectuer une analyse densitométrique de la phospho-CaMKII normalisée par rapport à la CaMKII totale. Comparer les conditions d'illumination et de non-illumination afin de quantifier l'efficacité de la photoactivation.
Mesures de contrôle de la qualité
Contrôle obscur : Les cellules chargées de NP-EGTA mais non éclairées établissent une phosphorylation de base de CaMKII.
Contrôle par la lumière seule : les cellules exposées à la lumière UV sans NP-EGTA confirment que l’illumination seule n’active pas la CaMKII.
Contrôle de la chélation du calcium : L’ajout de chélateurs de calcium rapides tels que le BAPTA confirme la dépendance de la signalisation au calcium.
Contrôle du chargement : Des niveaux constants de CaMKII total et de protéines de ménage assurent un chargement protéique égal.
Réplicats : Effectuer au moins trois réplicats biologiques indépendants.
Interprétation des résultats
Modèle de résultats attendus
-
Les cellules chargées en NP-EGTA sans illumination présentent de faibles niveaux de phospho-CaMKII.
-
Les cellules chargées de NP-EGTA illuminées par UV présentent une augmentation rapide et transitoire de la phospho-CaMKII.
-
La quantité totale de CaMKII reste inchangée quelles que soient les conditions.
Ce schéma indique une photoactivation réussie de la signalisation CaMKII dépendante du Ca²⁺.
Résultats alternatifs et interprétation
-
L'absence d'augmentation de la phospho-CaMKII peut indiquer une exposition insuffisante à la lumière, un chargement incomplet du calcium piégé ou une forte inhibition par des chélateurs endogènes.
-
L'activation prolongée de CaMKII peut refléter une libération excessive de calcium ou une activité phosphatase altérée.
Conclusion
Les approches décrites offrent des moyens précis et complémentaires de moduler la signalisation cellulaire. Chaque méthode permet un contrôle unique de l'activité des voies de signalisation, depuis la pharmacologie dose-dépendante jusqu'à l'activation rapide, réversible ou photo-induite, permettant ainsi d'analyser la dynamique et la causalité de la signalisation. Associés à des contrôles rigoureux et à une analyse quantitative, ces outils permettent de mieux comprendre les mécanismes de transduction du signal cellulaire et soutiennent la conception rationnelle d'interventions ciblées, tant dans le cadre de la recherche que dans celui de la thérapie.