Chromatographie liquide à haute performance couplée à la spectrométrie de masse

Chromatographie Liquide à Haute Performance couplée à la Spectrométrie de Masse (CLHP-MS) Protocole
Introduction
La chromatographie liquide à haute performance couplée à la spectrométrie de masse (CLHP-MS ou LC-MS) représente l’une des techniques analytiques les plus puissantes et polyvalentes de la chimie analytique moderne (Ardrey, 2003). Cette technique couplée combine les capacités de séparation de la chromatographie liquide à haute performance avec la puissance de détection et les capacités d’élucidation structurale de la spectrométrie de masse, permettant l’identification et la quantification de mélanges complexes avec une sensibilité et une sélectivité sans précédent (Niessen, 2006).
Le couplage de la CLHP avec la MS surmonte les limitations de chacune des techniques prises isolément. Alors que la CLHP offre une excellente séparation des mélanges complexes, ses systèmes de détection (UV-Vis, fluorescence et indice de réfraction) manquent souvent de spécificité et de sensibilité requises pour l’analyse de traces dans des matrices complexes (Snyder et al., 2010). La spectrométrie de masse, quant à elle, offre une sensibilité exceptionnelle et une spécificité moléculaire, mais nécessite une séparation préalable des mélanges complexes pour éviter la suppression ionique et la complexité spectrale (de Hoffmann et Stroobant, 2007). La combinaison synergique de ces techniques a révolutionné des domaines tels que l’analyse pharmaceutique, la protéomique, la métabolomique, la surveillance environnementale, la sécurité alimentaire et le diagnostic clinique (Gika et al., 2014).
Principe de la CLHP-MS
1. Principes de la chromatographie liquide à haute performance
La CLHP sépare les composés en fonction de leur distribution différentielle entre une phase mobile (solvant liquide) et une phase stationnaire (support solide tassé dans une colonne) (Guiochon et Guillemin, 1988). L’équation fondamentale régissant la séparation chromatographique est l’équation de van Deemter, qui relie la hauteur équivalente à un plateau théorique (HETP, H) à la vitesse linéaire de la phase mobile (u) (van Deemter et al., 1956) :
H = A + B/u + Cu
Où :
- A représente la diffusion turbulente (chemins d’écoulement multiples)
- B représente la diffusion longitudinale
- C représente la résistance au transfert de masse
L’efficacité de la séparation est inversement proportionnelle à la HETP ; des valeurs de H plus faibles indiquent une meilleure séparation (Giddings, 1965). Le nombre de plateaux théoriques (N) dans une colonne est calculé comme suit :
N = L/H = 5,54(tR/w1/2)²
Où L est la longueur de la colonne, tR est le temps de rétention et w1/2 est la largeur du pic à mi-hauteur (Neue, 1997).
Le temps de rétention d’un analyte en CLHP dépend de son coefficient de distribution (K) entre les phases mobile et stationnaire (Poole, 2003) :
K = Cs/Cm
Où Cs est la concentration dans la phase stationnaire et Cm est la concentration dans la phase mobile. Le facteur de rétention (k’) est lié à K par :
k’ = K(Vs/Vm)
Où Vs est le volume de phase stationnaire et Vm est le volume de phase mobile.
La résolution (Rs) entre deux pics est définie comme :
Rs = 2(tR2 - tR1)/(w1 + w2)
Où tR1 et tR2 sont les temps de rétention des pics 1 et 2, et w1 et w2 sont leurs largeurs respectives à la base. Une résolution ≥ 1,5 indique une séparation jusqu’à la ligne de base.
La résolution peut être reliée à l’efficacité de la colonne (N), à la sélectivité (α) et au facteur de rétention (k’) par l’équation fondamentale de résolution :
Rs = (√N/4) × [(α-1)/α] × [k’/(1+k’)]
Cette équation montre que la résolution peut être améliorée en augmentant l’efficacité de la colonne, en optimisant la sélectivité ou en ajustant la rétention.
Les différents modes de CLHP exploitent divers mécanismes de rétention :
- CLHP en phase inversée (CLHP-RP) : Mode le plus courant, utilisant une phase stationnaire non polaire (généralement silice greffée C18, C8 ou phényle) et une phase mobile polaire (mélanges eau-solvant organique). La rétention est principalement due aux interactions hydrophobes ; les composés les plus hydrophobes sont retenus plus longtemps.
- CLHP en phase normale (CLHP-NP) : Utilise une phase stationnaire polaire (silice, amino, cyano) et une phase mobile non polaire (hexane, chloroforme). Les composés polaires sont retenus plus fortement par liaisons hydrogène et interactions dipôle-dipôle.
- Chromatographie d’échange d’ions : Sépare les molécules chargées en fonction des interactions électrostatiques avec des groupes fonctionnels chargés sur la phase stationnaire. Les échangeurs de cations ont des groupes négativement chargés, les échangeurs d’anions des groupes positivement chargés.
- Chromatographie d’exclusion stérique (SEC) : Sépare les molécules selon leur volume hydrodynamique ; les molécules les plus grosses sont éluées en premier. Utilisée couramment pour l’analyse des protéines et polymères.
- Chromatographie d’interaction hydrophile (HILIC) : Variante de la chromatographie en phase normale utilisant des phases stationnaires polaires avec des phases mobiles aqueuses-organiques, particulièrement utile pour les composés polaires et hydrophiles.
2. Principes de la spectrométrie de masse
Un spectromètre de masse comporte trois composants fondamentaux : une source d’ionisation, un analyseur de masse et un détecteur. La séquence opérationnelle de base comprend (Figure 1) :
- Ionisation : conversion des molécules neutres en ions en phase gazeuse
- Analyse de masse : séparation des ions selon leur rapport masse/charge (m/z)
- Détection : mesure de l’abondance des ions à chaque valeur de m/z
Le spectre de masse résultant représente l’abondance ionique en fonction du m/z, fournissant une empreinte moléculaire de l’analyte.
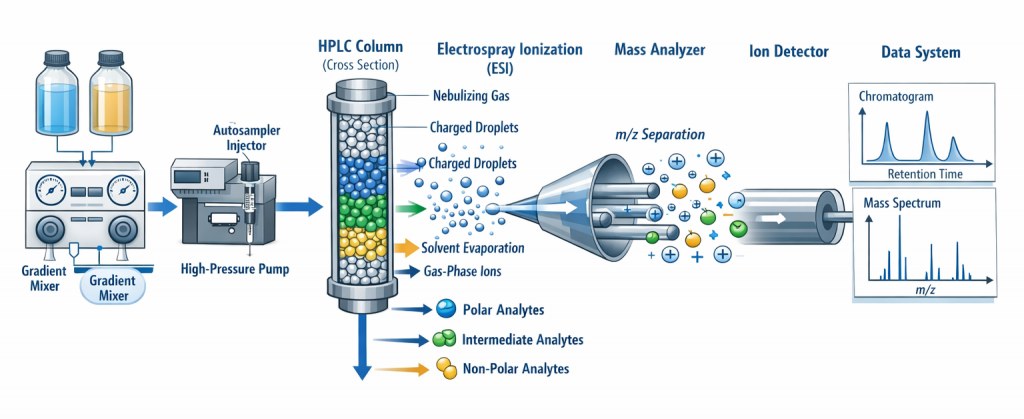
Figure 1 : Principe de la chromatographie liquide à haute performance couplée à la spectrométrie de masse (CLHP-MS ou LC-MS)
1. Préparation avant analyse
La préparation des échantillons est la première étape la plus critique en CLHP-MS, représentant jusqu’à 80 % du temps d’analyse et des sources d’erreurs, car elle impacte directement la sensibilité, la sélectivité et les effets de matrice (suppression/amplification ionique). Une mauvaise préparation peut entraîner un encrassement de la colonne, une variabilité du signal ou des faux négatifs/positifs. Viser un taux de récupération >90 % et une déviation standard relative (DSR) <15 % sur les répliques. Les objectifs principaux de la préparation des échantillons sont :
- Extraction et concentration de l’analyte
- Élimination des composants de la matrice (protéines, sels, lipides)
- Stabilisation et conservation de l’analyte
- Compatibilité du solvant avec la phase mobile de CLHP
Une mauvaise préparation des échantillons peut entraîner :
- Suppression ou amplification ionique
- Contamination de la colonne et réduction de sa durée de vie
- Effet mémoire entre les injections
- Formes de pics dégradées et mauvaise résolution
- Résultats non reproductibles
1.1. Préparation des échantillons de plasma et sérum
1.1.1. Précipitation des protéines
C’est la méthode la plus simple et la plus utilisée pour éliminer les protéines des matrices biologiques.
- Matériel
- Échantillons de plasma ou de sérum
- Acétonitrile (qualité CLHP-MS)
- Solution d’étalon interne
- Tubes à microcentrifugation de 1,5 mL
- Agitateur vortex
- Centrifugeuse
- Filtres seringue 0,2 µm ou plaques filtres
- Procédure
1. Décongélation : Retirer les échantillons du stockage à -80 °C et les décongeler à température ambiante (20-25 °C) pendant 30 minutes. Vortexer brièvement pour assurer l’homogénéité.
2. Aliquotage : Transférer 100 µL de plasma/sérum dans un tube à microcentrifugation de 1,5 mL à l’aide d’une pipette calibrée.
3. Addition de l’étalon interne : Ajouter 10 µL de solution d’étalon interne (typiquement 1-10 µg/mL dans du méthanol ou de l’acétonitrile). Vortexer 10 secondes.
4. Précipitation des protéines : Ajouter 300 µL d’acétonitrile glacé (rapport 3:1) à l’échantillon. Le rapport optimal peut varier selon la teneur en protéines :
- Plasma/sérum : 3:1 à 4:1 (organique:échantillon)
- Sang total : 4:1 à 5:1
- Homogénat de tissu : 5:1 à 10:1
5. Mélange : Vortexer vigoureusement pendant 1 minute pour assurer une précipitation complète des protéines.
6. Centrifugation : Centrifuger à 15 000 × g pendant 10 minutes à 4 °C pour peler les protéines précipitées.
7. Transfert du surnageant : Transférer soigneusement le surnageant dans un tube propre, en évitant le culot protéique. Typiquement 300-350 µL sont récupérés.
8. Évaporation (optionnelle) : Si une concentration est nécessaire, évaporer le surnageant à sec sous azote à 40 °C à l’aide d’un concentrateur d’échantillons. Reconstituer dans 100 µL de la phase mobile initiale.
9. Filtration : Filtrer le surnageant à travers un filtre seringue nylon ou PTFE de 0,2 µm dans un flacon CLHP.
10. Stockage : Conserver les échantillons préparés à 4 °C et les analyser dans les 24 heures, ou à -20 °C pour un stockage plus long.
1.1.2. Extraction en phase solide (SPE)
La SPE offre un nettoyage et une sélectivité supérieurs à la précipitation des protéines.
- Matériel
- Échantillons de plasma ou de sérum
- Cartouches SPE
- Solvant de conditionnement : méthanol (qualité CLHP-MS)
- Solvant d’équilibrage : eau (qualité CLHP-MS)
- Solvant de lavage : 5 % de méthanol dans l’eau
- Solvant d’élution : méthanol ou acétonitrile
- Manifold à vide SPE
- Concentrateur d’échantillons (évaporateur azote ou centrifugeuse sous vide)
- Procédure
1. Préparation de l’échantillon : Diluer 100 µL de plasma avec 300 µL d’eau contenant 0,1 % d’acide formique. Ajouter l’étalon interne.
2. Conditionnement de la cartouche :
- Faire passer 1 mL de méthanol à travers la cartouche à 1-2 mL/min
- Ne pas laisser sécher la cartouche
3. Équilibrage de la cartouche :
- Faire passer 1 mL d’eau à travers la cartouche à 1-2 mL/min
- Maintenir l’humidité du lit sorbant
4. Chargement de l’échantillon :
- Charger l’échantillon dilué sur la cartouche à 1 mL/min
- Recueillir l’effluent si un test de récupération est nécessaire
5. Lavage :
- Laver avec 1 mL de 5 % de méthanol dans l’eau
- Cela élimine les sels et les composants polaires de la matrice tout en retenant les analytes
6. Séchage de la cartouche :
- Appliquer un vide complet pendant 5 minutes pour éliminer le solvant de lavage résiduel
- Cette étape est critique pour une élution efficace
7. Élution :
- Éluer les analytes avec 500 µL de méthanol (ou d’acétonitrile pour les composés plus hydrophobes)
- Recueillir l’éluat dans un tube propre
- Pour une meilleure récupération, utiliser deux aliquots de 250 µL plutôt qu’un seul de 500 µL
8. Évaporation :
- Évaporer à sec sous azote à 40 °C
- Reconstituer dans 100 µL de la phase mobile initiale
9. Filtration et analyse :
- Filtrer si nécessaire et transférer dans un flacon CLHP
- Analyser immédiatement ou conserver à 4 °C
1.2. Préparation des échantillons d’urine
L’urine est généralement une matrice plus propre que le plasma mais nécessite une dilution pour réduire la teneur en sels.
- Dilution directe avec étalon interne (« dilute-and-shoot »)
- Matériel
- Échantillons d’urine
- Eau (qualité CLHP-MS)
- Solution d’étalon interne
- Filtres seringue 0,2 µm
- Procédure
1. Décongélation : Décongeler les échantillons d’urine à température ambiante et mélanger par inversion.
2. Centrifugation : Centrifuger à 3 000 × g pendant 10 minutes pour éliminer les particules.
3. Dilution : Diluer l’urine 1:10 (v/v) avec de l’eau contenant l’étalon interne
- Exemple : 50 µL d’urine + 450 µL d’eau avec étalon interne
4. Filtration : Filtrer à travers un filtre seringue 0,2 µm dans un flacon CLHP.
5. Analyse : Analyser immédiatement ou conserver à 4 °C jusqu’à 24 heures.
1.3. Préparation des échantillons de tissus
- Homogénéisation et extraction de tissus
- Matériel
- Échantillons de tissus
- Tampon phosphate salin (PBS), pH 7,4
- Acétonitrile ou méthanol (qualité CLHP-MS)
- Solution d’étalon interne
- Homogénéiseur de tissus
- Centrifugeuse réfrigérée
- Procédure
1. Pesée du tissu : Peser précisément 50-100 mg de tissu.
2. Homogénéisation :
- Ajouter 3-5 volumes de PBS glacé (ex. : 300-500 µL pour 100 mg de tissu)
- Ajouter l’étalon interne
- Homogénéiser à l’aide d’un homogénéiseur
- Maintenir les échantillons sur glace tout au long du processus
3. Précipitation des protéines :
- Ajouter 3 volumes d’acétonitrile glacé à l’homogénat
- Vortexer 1 minute
4. Centrifugation : Centrifuger à 15 000 × g pendant 15 minutes à 4 °C.
5. Collecte du surnageant : Transférer le surnageant dans un tube propre.
6. Évaporation (optionnelle) : Évaporer à sec et reconstituer si une concentration est nécessaire.
7. Filtration : Filtrer à travers un filtre 0,2 µm et transférer dans un flacon CLHP.
1.4. Préparation des échantillons pour résidus de pesticides dans les aliments
- Méthode QuEChERS modifiée
- Matériel
- Échantillon alimentaire homogénéisé
- Acétonitrile avec 1 % d’acide acétique
- Sels d’extraction QuEChERS (4 g MgSO₄, 1 g NaCl, 1 g citrate de sodium, 0,5 g sesquihydrate de citrate disodique)
- Nettoyage SPE dispersif (PSA, C18, GCB)
- Procédure
1. Homogénéisation de l’échantillon : Homogénéiser 10-15 g d’échantillon alimentaire au mixeur.
2. Extraction :
- Peser 10 g d’échantillon homogénéisé dans un tube à centrifuger de 50 mL
- Ajouter 10 mL d’acétonitrile avec 1 % d’acide acétique
- Ajouter les étalons internes
- Agiter vigoureusement 1 minute
3. Addition des sels :
- Ajouter le sachet de sels d’extraction QuEChERS
- Agiter immédiatement 1 minute
- Centrifuger à 4 000 × g pendant 5 minutes
4. Nettoyage :
- Transférer 6 mL de surnageant dans un tube SPE dispersif contenant :
* 900 mg MgSO₄
* 150 mg PSA (élimine acides organiques, sucres)
* 150 mg C18 (élimine lipides, cires)
* 45 mg GCB (carbone graphité, élimine pigments)
- Vortexer 30 secondes
- Centrifuger à 4 000 × g pendant 5 minutes
5. Filtration et analyse :
- Filtrer 1 mL de surnageant à travers un filtre 0,2 µm
- Transférer dans un flacon CLHP et analyser
1.5. Préparation des échantillons pharmaceutiques
- Extraction et analyse de comprimés
- Matériel
- Comprimés pharmaceutiques
- Diluant : méthanol ou mélange acétonitrile/eau
- Bain ultrasonore
- Fioles jaugées
- Procédure
1. Pesée des comprimés : Peser précisément 10 comprimés et calculer le poids moyen.
2. Broyage : Réduire les comprimés en poudre fine à l’aide d’un mortier et pilon.
3. Pesée : Peser la poudre équivalente à un comprimé dans une fiole jaugée.
4. Extraction :
- Ajouter 70 % du volume final de diluant
- Soniquer 15 minutes
- Refroidir à température ambiante
- Compléter au volume avec le diluant
5. Filtration : Filtrer à travers un filtre 0,45 µm.
6. Dilution : Diluer à la concentration appropriée pour l’analyse CLHP-MS (typiquement 1-100 µg/mL).
7. Analyse : Analyser par rapport à des solutions d’étalons de référence.
2. Développement et optimisation de la méthode
2.1. Développement de la méthode CLHP
2.1.1 Stratégie de sélection de la colonne
Le choix de la phase stationnaire est la deuxième décision la plus critique dans le développement d’une méthode CLHP. Les critères de sélection comprennent :
a. Propriétés de l’analyte
Log P (coefficient de partage octanol-eau) : Indique l’hydrophobicité
- Log P < 0 : Très hydrophile → HILIC ou échange d’ions
- Log P 0-3 : Modérément hydrophile → C18 avec phase mobile aqueuse
- Log P > 3 : Hydrophobe → C18, C8 ou phényle
pKa : Détermine l’état d’ionisation à différents pH
- Pour les acides : utiliser pH > pKa + 2 pour une ionisation complète
- Pour les bases : utiliser pH < pKa - 2 pour une ionisation complète
- Ou utiliser un pH où l’analyte est neutre pour une rétention en phase inversée
Masse moléculaire : Les grosses molécules peuvent nécessiter des tailles de pores plus grandes
- <1 000 Da : taille de pore 100 Å
- 1 000-10 000 Da : 200-300 Å
- >10 000 Da : 300-500 Å
2.1.2 Optimisation de la phase mobile
a. Sélection des solvants
Les combinaisons de phases mobiles les plus courantes en CLHP-MS sont :
a.1. Eau/Acétonitrile (A/B):
- Combinaison la plus populaire
- Bonne compatibilité MS
- Adaptée à la plupart des applications en phase inversée
- Viscosité inférieure à eau/méthanol
- Alternative à l’acétonitrile
- Meilleure pour les composés très hydrophobes
- Pression arrière plus élevée en raison de la viscosité supérieure
a.3. Acétonitrile/Eau (HILIC) (A/B):
- Teneur élevée en organique (>60 %)
- Pour les composés polaires et hydrophiles
b. Sélection des additifs compatibles avec la MS
Les additifs de phase mobile servent à contrôler le pH, au pairage ionique et à l’amélioration de l’ionisation.
b.1. Acides volatils (mode ion positif)
- Acide formique (0,1-0,5 %) : le plus courant, pKa = 3,75
- Favorise la protonation en ESI
- Améliore la forme des pics pour les composés basiques
- Concentration typique : 0,1 % (v/v)
- Acide acétique (0,1-0,5 %) : acide plus faible, pKa = 4,76
- Moins de suppression ionique que l’acide formique
- Meilleur pour les composés labiles
b.2. Bases volatiles (mode ion négatif)
- Hydroxyde d’ammonium (0,01-0,1 %) : pKa = 9,25
- Favorise la déprotonation des composés acides
- Concentration typique : 0,01 % (v/v)
b.3. Tampons volatils
- Formiate d’ammonium (2-20 mM) : pH 3-4
- Fournit un tamponnage de pH avec compatibilité MS
- Concentration typique : 5 mM
- Acétate d’ammonium (2-20 mM) : pH 4-7
- Plage de tamponnage plus large
- Bon pour les composés neutres et faiblement ionisables
b.4. Additifs non volatils (à éviter en MS)
- Tampons phosphate : provoquent une suppression ionique et une contamination
- Acide trifluoroacétique (TFA) : fort pairage ionique, suppression sévère du signal
- Sels non volatils : contaminent la source d’ions
2.1.3 Optimisation du gradient
L’élution en gradient est préférable à l’élution isocratique pour les mélanges complexes, offrant une meilleure capacité de pics et un temps d’analyse plus court.
a. Screening initial du gradient
Un gradient générique pour le développement de méthode
| Temps (min) | %B (organique) |
| 0 | 5 |
| 1 | 5 |
| 11 | 95 |
| 13 | 95 |
| 13,1 | 5 |
| 15 | 5 |
b. Stratégies d’optimisation du gradient
1. Gradients segmentés : gradients doux dans les zones d’intérêt, gradients raides là où aucun pic n’élue.
2. Gradients par paliers : paliers isocratiques suivis d’augmentations rapides, utiles pour les composés de polarités très différentes.
3. Optimisation de la pente du gradient :
- Calculer la pente du gradient (Δ%B/min)
- Ajuster pour obtenir Rs > 1,5 entre les paires critiques
- Pentes typiques : 1-5 %B/min pour les mélanges complexes
2.1.4 Optimisation de la température
La température de la colonne influence la rétention, la sélectivité et l’efficacité.
a. Effets de la température
a.1. Augmentation de température :
- Diminution de la rétention (k’ plus faible)
- Diminution de la viscosité → pression arrière plus faible
- Augmentation du transfert de masse → meilleure efficacité
- Changements possibles de sélectivité
a.2. Stratégie d’optimisation :
- Tester les températures : 25, 35, 45, 55 °C
- Plage typique : 30-50 °C
- Températures plus élevées (60-80 °C) pour les composés très hydrophobes
a.2. Analyse de van’t Hoff :
La relation entre température et rétention est décrite par :
ln k’ = -ΔH°/RT + ΔS°/R + ln ϕ
Où ΔH° est l’enthalpie, ΔS° est l’entropie, R est la constante des gaz, T est la température et ϕ est le rapport de phases.
2.2 Optimisation de la spectrométrie de masse
2.2.1 Optimisation de la source d’ionisation
a. Paramètres d’ionisation par électrospray (ESI)
a.1. Tension de pulvérisation (tension capillaire) :
- Mode positif : +2 500 à +4 500 V
- Mode négatif : -2 000 à -3 500 V
- Optimiser pour un signal maximal sans décharge
a.2. Pression du gaz nébuliseur :
- Plage typique : 20-60 psi (azote)
- Pression plus élevée : gouttelettes plus petites, désolvatation plus efficace
- Optimiser pour chaque débit
a.3. Débit du gaz de séchage :
- Plage typique : 5-15 L/min (azote)
- Débit plus élevé : meilleure désolvatation mais risque de perte d’ions
- Augmenter avec le débit et la teneur aqueuse
a.4. Température du gaz de séchage :
- Plage typique : 200-350 °C
- Température plus élevée : meilleure désolvatation
- Éviter une chaleur excessive pour les composés thermolabiles
a.5. Gaz de gaine :
- Débit : 5-12 L/min
- Température : 200-400 °C
- Améliore la sensibilité pour certains composés
b. Paramètres APCI
b.1. Température du vaporiseur :
- Plage typique : 300-500 °C
- Doit être suffisamment élevée pour une vaporisation complète
- Optimiser selon la stabilité thermique du composé
b.2. Courant de décharge corona :
- Plage typique : 2-8 µA
- Courant plus élevé : plus d’ionisation mais plus de bruit
b.3. Gaz nébuliseur et de séchage :
- Similaires à l’ESI mais tolèrent généralement des températures plus élevées
2.2.2 Optimisation de l’analyseur de masse
a. Paramètres du quadrupôle
a.1. Résolution :
- Résolution unitaire : 0,7 Da FWHM à m/z 609
- Optimiser pour l’analyse quantitative (résolution unitaire) ou la sélectivité (résolution étroite)
a.2. Temps de séjour (pour SRM) :
- Plage typique : 5-100 ms par transition
- Temps de séjour plus long : meilleure sensibilité mais moins de points par pic
- Viser ≥10 points par pic
a.3. Énergie de collision (pour MS/MS) :
- Optimiser pour chaque transition précurseur → produit
- Point de départ typique : CE = 0,03 × m/z + 5 (V)
- Ajuster finement pour une intensité maximale de l’ion produit
b. Paramètres du temps de vol (TOF)
b.1. Résolution de masse :
- Typique : 10 000-40 000 FWHM
- Résolution plus élevée : meilleure précision de masse mais sensibilité plus faible
b.2. Taux d’acquisition :
- Typique : 1-20 spectres/seconde
- Équilibrer sensibilité et points de données par pic
b.3. Étalonnage de masse :
- Utiliser une correction de masse de référence pour une masse exacte
- Masses de référence typiques : m/z 121,0509 ; 922,0098 (purine, HP-0921)
c. Paramètres de l’Orbitrap
c.1. Résolution :
- Plage : 15 000-480 000 FWHM
- Résolution plus élevée : temps de balayage plus long
- Typique en métabolomique : 60 000-120 000
c.2. Cible AGC (Automatic Gain Control) :
- Contrôle la population d’ions dans le piège
- Typique : 1×10⁵ à 1×10⁶
- AGC plus élevé : meilleures statistiques mais temps de remplissage plus long
c.3. Temps d’injection maximal :
- Typique : 50-200 ms
- Équilibrer sensibilité et cycle de service
2.2.3 Optimisation de la MS en tandem
Le SRM/MRM est l’étalon-or pour l’analyse quantitative CLHP-MS/MS.
a. Flux de travail d’optimisation
a.1. Sélection de l’ion précurseur :
- Infuser un étalon pur (1-10 µg/mL) à 5-10 µL/min
- Acquérir un spectre MS balayage complet
- Identifier [M+H]⁺ (mode positif) ou [M-H]⁻ (mode négatif)
- Noter les adduits : [M+Na]⁺, [M+NH₄]⁺, [M+2H]²⁺
a.2. Balayage des ions produits :
- Sélectionner l’ion précurseur
- Balayer les ions produits sur une large plage (ex. m/z 50-500)
- Faire varier l’énergie de collision pour identifier les fragments optimaux
a.3. Sélection des transitions :
- Sélectionner l’ion produit le plus abondant pour la quantification (quantificateur)
- Sélectionner 1-2 ions produits supplémentaires pour la confirmation (qualificateurs)
- S’assurer que les transitions sont spécifiques (pas d’interférences)
a.4. Optimisation de l’énergie de collision :
- Tester la CE par incréments de 5 V
- Tracer l’intensité de l’ion produit en fonction de la CE
- Sélectionner la CE à l’intensité maximale
a.5. Optimisation du fragmentor/potentiel de déclustering :
- Optimiser pour une transmission maximale de l’ion précurseur
- Plage typique : 50-200 V
- Éviter une fragmentation excessive dans la source
b. Critères de qualité pour les transitions SRM
- Quantificateur : transition la plus abondante et spécifique
- Qualificateur : transitions secondaires pour confirmation
- Rapport d’ions : rapport qualificateur/quantificateur constant (±20 %)
2.3 Paramètres de validation de la méthode
La validation de méthode garantit que les procédures analytiques conviennent à leur usage prévu. Les lignes directrices réglementaires incluent ICH Q2(R1), FDA Bioanalytical Method Validation et les lignes directrices de l’EMA.
2.3.1 Spécificité/Sélectivité
- Définition
Capacité à mesurer l’analyte sans ambiguïté en présence d’autres composants.
- Procédure
- Analyser des échantillons de matrice blanche provenant d’au moins 6 sources différentes
- Analyser la matrice blanche enrichie avec l’analyte au LLOQ
- Analyser la matrice blanche enrichie avec des composés interférents potentiels
- S’assurer d’aucune interférence au temps de rétention de l’analyte (réponse <20 % du LLOQ)
2.3.2 Linéarité et domaine
- Définition
Capacité à obtenir des résultats directement proportionnels à la concentration de l’analyte.
- Procédure
- Préparer des étalons de calibration à 6-8 niveaux de concentration
- Analyser en triplicate sur trois jours distincts
- Construire la courbe de calibration avec le modèle de régression approprié :
- Linéaire : y = mx + b
- Quadratique : y = ax² + bx + c
- Linéaire pondérée : pondération 1/x ou 1/x²
- Critères d’acceptation
- Coefficient de corrélation (r²) ≥ 0,99
- Concentrations recalculées dans ±15 % de la valeur nominale (±20 % au LLOQ)
- Au moins 75 % des étalons respectent les critères
2.3.3 Exactitude et précision
- Exactitude : proximité de la valeur mesurée à la valeur vraie (% biais)
- Précision : degré d’accord entre les mesures répétées (% DSR)
- Procédure
- Préparer des échantillons QC à 3-4 niveaux : LLOQ, bas, moyen, élevé
- Analyser 5 répliques par niveau en une journée (intra-jour)
- Analyser sur 3 jours distincts (inter-jour)
- Calculer l’exactitude (% biais) et la précision (% DSR)
- Critères d’acceptation
- Exactitude : ±15 % de la valeur nominale (±20 % au LLOQ)
- Précision : DSR ≤15 % (≤20 % au LLOQ)
2.3.4 Sensibilité (LLOQ et LOD)
- Limite inférieure de quantification (LLOQ)
Concentration la plus faible avec exactitude et précision acceptables
- Critères d’acceptation pour le LLOQ
- Rapport signal/bruit ≥10
- Exactitude : 80-120 % de la valeur nominale
- Précision : DSR ≤20 %
- Limite de détection (LOD)
Concentration la plus faible détectable mais non nécessairement quantifiable
- Calcul
LOD = 3,3 × (DS de la réponse / pente de la courbe de calibration)
2.3.5 Effets de matrice
- Définition
Effet des composants de matrice co-élués sur l’ionisation de l’analyte.
- Procédure (méthode d’addition post-extraction)
- Préparer l’ensemble A : étalons purs dans la phase mobile
- Préparer l’ensemble B : extrait de matrice blanche enrichi après extraction
- Préparer l’ensemble C : matrice blanche enrichie avant extraction
- Analyser tous les ensembles (n=6 chacun)
- Calculer l’effet de matrice et la récupération
- Calculs
Effet de matrice (%) : (aire moyenne B / aire moyenne A) × 100
Récupération (%) : (aire moyenne C / aire moyenne B) × 100
Efficacité du procédé (%) : (aire moyenne C / aire moyenne A) × 100
- Critères d’acceptation
- Effet de matrice : 85-115 % (suppression/amplification minimale)
- Précision : DSR ≤15 %
2.3.6 Stabilité
- Définition
Stabilité chimique de l’analyte dans diverses conditions de stockage et de traitement.
- Types d’études de stabilité
1. Stabilité de la solution mère
- Stocker dans les conditions prévues (-20 °C, 4 °C, température ambiante)
- Tester à 0, 1, 3, 6 mois
- Comparer à une solution fraîchement préparée
2. Stabilité aux cycles de congélation-décongélation
- Congeler à la température de stockage prévue
- Décongeler à température ambiante
- Répéter 3 cycles
- Analyser et comparer aux échantillons frais
3. Stabilité sur paillasse
- Laisser les échantillons à température ambiante pendant le temps de traitement prévu
- Typique : 4-24 heures
- Analyser et comparer aux échantillons frais
4. Stabilité dans l’autosampler
- Stocker les échantillons préparés dans l’autosampler à la température de fonctionnement
- Typique : 24-72 heures
- Réanalyser et comparer à l’analyse initiale
5. Stabilité à long terme
- Stocker à la température de stockage prévue (ex. -80 °C)
- Tester à 1, 3, 6, 12 mois
- Comparer aux échantillons frais
- Critères d’acceptation
- Concentration de l’analyte dans ±15 % de la valeur nominale
- Échantillons considérés stables dans les conditions testées
3. Acquisition et traitement des données
3.1 Stratégies d’acquisition des données
3.1.1 Analyse quantitative ciblée (SRM/MRM)
a. Paramètres de la méthode
1. Type de balayage : Surveillance de réaction sélectionnée (SRM) ou surveillance de réactions multiples (MRM)
2. Transitions : Définir les paires ion précurseur → ion produit pour chaque analyte
- Transition quantificatrice : la plus abondante et spécifique
- Transitions qualificatrices : 1-2 supplémentaires pour confirmation
3. Temps de séjour : Temps consacré à chaque transition
- Typique : 10-50 ms
- Calcul : temps de cycle / nombre de transitions
- Viser ≥10 points de données par pic
4. Fenêtres de temps de rétention : Limiter les transitions à des fenêtres temporelles spécifiques
- Réduit le temps de cycle, augmente la sensibilité
- Typique : ±0,5 min autour du temps de rétention attendu
3.1.2 Métabolomique/lipidomique non ciblée
a. Balayage MS complet
a.1. Plage de balayage : suffisamment large pour capturer tous les analytes d’intérêt
- Typique : m/z 50-1 200 pour petites molécules
- m/z 100-2 000 pour lipides
a.2. Résolution : équilibre entre sensibilité et précision de masse
- Orbitrap : 60 000-120 000 FWHM
- TOF : 20 000-40 000 FWHM
a.3. Commutation de polarité : acquérir les modes positif et négatif
- Augmente la couverture de l’espace chimique
- Nécessite une commutation rapide de polarité (<100 ms)
b. Acquisition dépendante des données (DDA)
1. Balayage de surveillance : balayage MS complet pour identifier les précurseurs
2. Sélection des précurseurs : N ions les plus abondants (ex. Top 5-10)
3. Acquisition MS/MS : fragmenter les précurseurs sélectionnés
4. Liste d’exclusion : exclure les ions précédemment fragmentés (exclusion dynamique)
- Durée : 10-30 secondes
- Évite l’échantillonnage répété des ions abondants
b. Acquisition indépendante des données (DIA)
1. Acquisition par fenêtres séquentielles : fragmenter tous les ions dans des fenêtres m/z séquentielles
- Largeur de fenêtre : 10-25 Da
- Chevauchement : 1 Da
2. Fragmentation de tous les ions (AIF) : fragmenter tous les ions simultanément
- Simple mais spectres complexes
- Déconvolution requise
3. SWATH-MS : implémentation spécifique de DIA sur instruments Sciex
- Fenêtres de 25 Da sur toute la plage de masse
- Couverture MS/MS exhaustive
3.2 Flux de travail de traitement des données
3.2.1 Analyse quantitative (SRM/MRM)
a. Intégration des pics
a.1. Intégration automatique
- Le logiciel détecte les pics selon un seuil signal/bruit
- Intègre l’aire sous la courbe
a.2. Revue manuelle
- Vérifier les limites d’intégration
- Contrôler les interférences
- Assurer une intégration cohérente sur tous les échantillons
a.3. Paramètres d’intégration
- Lissage : Gaussien ou Savitzky-Golay (5-9 points)
- Soustraction de ligne de base : linéaire ou polynomiale
- Seuil de détection des pics : S/N >3
b. Construction de la courbe de calibration
b.1. Tracé : rapport des aires des pics (analyte/étalon interne) en fonction de la concentration
b.2. Choix du modèle de régression :
- Linéaire : y = mx + b (le plus courant)
- Quadratique : y = ax² + bx + c (grande gamme dynamique)
- Puissance : y = axᵇ (réponse non linéaire)
b.3. Pondération :
- Sans pondération : importance égale à tous les points
- Pondération 1/x : met l’accent sur les faibles concentrations
- Pondération 1/x² : accentue davantage la partie basse
b.4. Critères d’acceptation :
- r ² ≥ 0,99
- Concentrations recalculées dans ±15 % de la valeur nominale (±20 % au LLOQ)
- Au moins 75 % des étalons valides
c. Calcul de la concentration
Céchantillon = (Aireéchantillon / AireIS) × (1 / pente) × Facteur de dilution
d. Contrôle qualité
d.1. Vérification des échantillons QC :
- Doivent être dans ±15 % de la valeur nominale
- Au moins 67 % des QC doivent être valides
d.2. Acceptation du lot :
- Courbe de calibration conforme
- QC conformes
- Tests de convenance du système conformes
3.2.2 Analyse qualitative (identification de métabolites)
Flux de travail pour l’identification d’inconnus :
1. Détection des pics :
- Extraire les chromatogrammes d’ions (EIC) pour toutes les valeurs m/z détectées
- Appliquer l’algorithme de détection de pics (détection centroïde)
- Seuil typique : S/N >5
2. Déconvolution :
- Séparer les pics co-élués
- Attribuer les spectres MS/MS aux précurseurs corrects
3. Recherche de masse exacte :
- Calculer la formule moléculaire à partir de la masse exacte
- Rechercher dans les bases de données (METLIN, HMDB, LIPID MAPS, PubChem)
- Tolérance de masse : ±5 ppm
4. Correspondance MS/MS :
- Comparer le spectre MS/MS expérimental aux spectres de bibliothèque
- Algorithmes de scoring : produit scalaire, similarité cosinus
- Seuil de correspondance : score >0,7
5. Niveaux de confiance (Metabolomics Standards Initiative) :
- Niveau 1 : confirmé avec étalon authentique
- Niveau 2 : annotation putative (correspondance bibliothèque)
- Niveau 3 : composé caractérisé par classe
- Niveau 4 : inconnu
3.3 Analyse statistique
3.3.1 Statistiques univariées
Changement de pli (fold change) :
- Rapport des intensités moyennes entre groupes
- Seuil : fold change >2 ou <0,5
Test t :
- Compare les moyennes entre deux groupes
- Hypothèse : distribution normale
- Significativité : p <0,05
ANOVA :
- Compare les moyennes entre ≥3 groupes
- Tests post-hoc : Tukey, Bonferroni
3.3.2 Statistiques multivariées
Analyse en composantes principales (ACP) :
- Réduction de dimension non supervisée
- Identifie les principales sources de variation
- Détecte les valeurs aberrantes
Analyse discriminante par moindres carrés partiels (PLS-DA) :
- Méthode de classification supervisée
- Maximise la séparation entre groupes prédéfinis
- Importance des variables : scores VIP >1
Regroupement hiérarchique :
- Regroupe les échantillons ou métabolites selon leur similarité
- Visualisation par dendrogramme
4. Références
Ardrey, R. E. (2003). Liquid Chromatography-Mass Spectrometry: An Introduction. John Wiley & Sons.
Niessen, W. M. A. (2006). Liquid Chromatography-Mass Spectrometry (3rd ed.). CRC Press.
Snyder, L. R., Kirkland, J. J., & Dolan, J. W. (2010). Introduction to Modern Liquid Chromatography (3rd ed.). John Wiley & Sons.
de Hoffmann, E., & Stroobant, V. (2007). Mass Spectrometry: Principles and Applications (3rd ed.). John Wiley & Sons.
Gika, H. G., Theodoridis, G. A., & Wilson, I. D. (2014). Liquid chromatography and ultra-performance liquid chromatography-mass spectrometry fingerprinting of human urine. Journal of Chromatography A, 1333, 41-48.
Guiochon, G., & Guillemin, C. L. (1988). Quantitative Gas Chromatography for Laboratory Analyses and On-Line Process Control.
van Deemter, J. J., Zuiderweg, F. J., & Klinkenberg, A. (1956). Longitudinal diffusion and resistance to mass transfer as causes of nonideality in chromatography. Chemical Engineering Science, 5(6), 271-289.
Giddings, J. C. (1965). Dynamics of Chromatography: Part I. Principles and Theory. Marcel Dekker.
Neue, U. D. (1997). HPLC Columns: Theory, Technology, and Practice. Wiley-VCH.
Poole, C. F. (2003). The Essence of Chromatography.