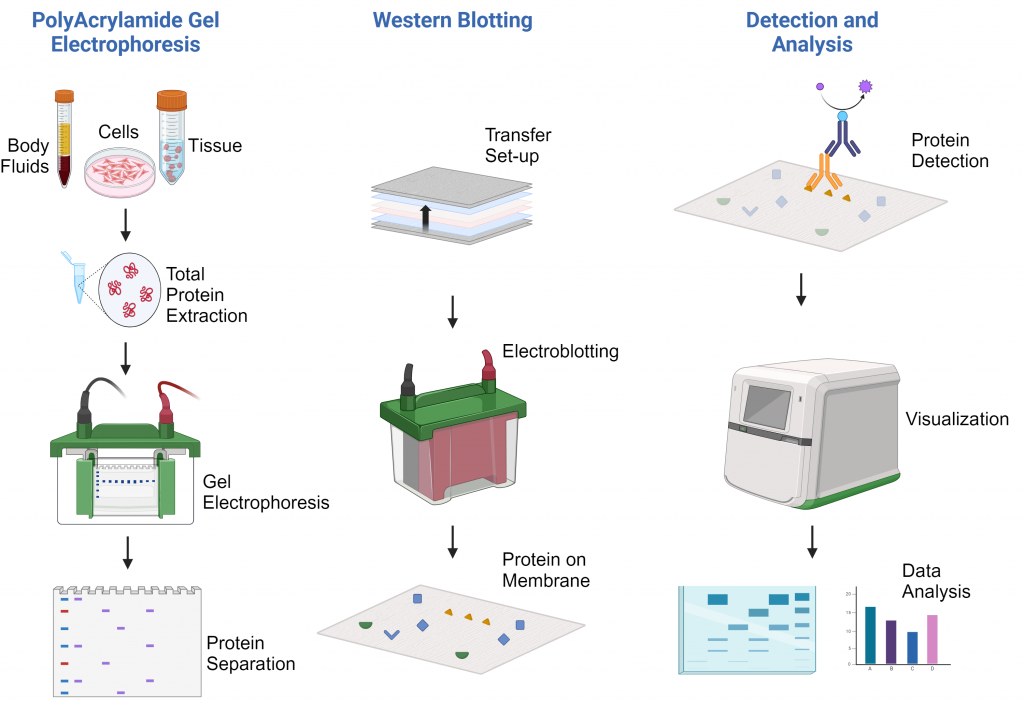
Western blot protocole

Principe de la technique
Le principe du Western blotting implique plusieurs étapes critiques :
-
Séparation des protéines : Les protéines sont séparées en fonction de leur taille par SDS-PAGE.
-
Transfert : Les protéines séparées sont transférées sur une membrane solide (nitrocellulose ou PVDF).
-
Blocage : Les surfaces membranaires sont bloquées pour empêcher toute liaison non spécifique.
-
Détection : Des anticorps primaires spécifiques reconnaissent les protéines cibles, suivis d'anticorps secondaires marqués.
-
Visualisation : Les complexes protéine-anticorps sont visualisés par des réactions chimiluminescentes, fluorescentes ou colorimétriques.
Le succès de la technique dépend fortement de la spécificité des anticorps et de l'intégrité de la préparation de l'échantillon.
La première étape importante à ne pas négliger lors de la réalisation d'un western blotting est la préparation de l'échantillon. Pour obtenir des résultats reproductibles, il convient d'extraire et de purifier efficacement les protéines à l'aide d'une méthode d'homogénéisation adaptée, capable de libérer efficacement le contenu intracellulaire de la cellule par la rupture des membranes cellulaires.
Isolation d'échantillons de protéines à partir de différents types d'échantillons
L'extraction et l'isolement des protéines sont les premières étapes critiques d'un large éventail de techniques biochimiques et de biologie moléculaire, notamment le transfert de Western, la spectrométrie de masse, les essais enzymatiques et l'immunoprécipitation. Le succès des applications en aval dépend fortement de la qualité, de l'intégrité et de la représentativité des échantillons de protéines isolées.
Les échantillons biologiques tels que les cellules cultivées, les tissus, les bactéries, les levures et le matériel végétal présentent chacun des défis uniques pour l'extraction des protéines. Par conséquent, le choix d'une stratégie d'isolement appropriée est essentiel pour maximiser le rendement, préserver la fonctionnalité des protéines et minimiser la dégradation ou la modification.
1. Homogenization
Choisissez une méthode d'homogénéisation en fonction du type d'échantillon :
-
Ultrasons (pour les échantillons délicats)
-
Presse française (pour les bactéries, les levures)
-
Broyage de billes de verre (pour les cellules coriaces, les champignons)
-
Homogénéisateur Dounce/Potter-Elvehjem (pour les tissus mous)
-
Broyage manuel à l'aide d'un mortier ou d'un pilon (pour les tissus congelés)
Objectif : lyse cellulaire efficace sans dégradation des protéines.
2. Manipulation des échantillons Post-traitement
Après une exposition aux xénobiotiques ou une manipulation génétique (par exemple, siRNA knockdown) :
- Congeler immédiatement les échantillons dans l'azote liquide
- Lyser immédiatement pour éviter l'activité des protéases endogènes.
Toujours garder les échantillons au froid pendant le traitement.
3. Gestion des cycles de gel/dégel
Éviter les cycles multiples de congélation/décongélation pour protéger l'intégrité des protéines.
Aliquoter les échantillons pour éviter toute décongélation inutile.
4. Sélection du tampon de lyse
a. Faire correspondre le tampon à l'emplacement de la protéine cible :
-
Protéines cytosoliques → tampon de lyse doux (par exemple, NP-40, Triton X-100)
-
Protéines liées à la membrane → tampons plus forts (par exemple, RIPA)
-
Protéines nucléaires/mitochondriales → tampons spécialisés (par exemple, tampon hypotonique, tampon d'homogénéisation pour drosophile)
b. Adapter le tampon à la sensibilité de l'anticorps :
-
Détection des protéines natives → Détergent doux/non ionique ou pas de détergent
-
Détection des protéines dénaturées → conditions fortes/dénaturantes
-
Inclure des inhibiteurs de protéase et de phosphatase dans tous les tampons de lyse.
c. Quelques produits courants et pourquoi ils sont utilisés :
-
HEPES : Maintient la stabilité du pH.
-
Saccharose / Mannitol : Stabilisateurs osmotiques pour éviter l'éclatement des organites.
-
EDTA / EGTA: chélate les cations divalents pour inhiber les métalloprotéases et empêcher la dégradation.
-
Inhibiteurs de Protéase et dephosphatase : Protègent les protéines de la dégradation enzymatique.
5. Notes critiques sur le fractionnement des échantillons
a. Attention à la perte de protéines lors de la centrifugation :
-
Tester si la protéine cible est retenue dans le culot (débris).
-
Exemple : Jusqu'à 50 % de la myosine et ~66 % de la CSQ2 ont été perdus dans les débris s'ils n'ont pas été contrôlés.
b. Il faut toujours vérifier si la protéine en question est :
-
Soluble (cytoplasmique)
-
Insoluble (cytosquelette)
-
Lié à la membrane (organellaire)
Séparation électrophorétique
Après l'extraction et la quantification des protéines, la phase critique suivante de l'analyse protéomique est la séparation électrophorétique. L'électrophorèse sur gel reste une technique fondamentale pour améliorer la sélectivité et la sensibilité de la recherche sur le protéome, en permettant la résolution de mélanges complexes de protéines sur la base de leurs propriétés physicochimiques.

Diverses matrices de gel - dont l'agarose, l'amidon et le polyacrylamide - ont été mises au point pour les applications électrophorétiques. Parmi celles-ci, le polyacrylamide est la matrice de choix pour l'analyse des protéines en raison de sa stabilité mécanique, de son inertie chimique et de la possibilité de réguler précisément la polymérisation avec le bisacrylamide. Un rapport acrylamide/bisacrylamide de 37,5:1, couramment utilisé, permet de générer des gels robustes avec des pores de taille reproductible, adaptés au fractionnement des protéines à haute résolution.
Deux formes principales d'électrophorèse sur gel de polyacrylamide (PAGE) sont largement utilisées : la PAGE native et la SDS-PAGE dénaturante.
-
La PAGE Native préserve les structures tertiaires et quaternaires des protéines, ce qui permet de les séparer sur la base d'une combinaison de charge nette, de taille hydrodynamique et de conformation. Dans les systèmes tampons alcalins, la plupart des protéines acquièrent une charge négative nette, ce qui leur permet de migrer vers l'anode sans dénaturation. Un avantage majeur de la PAGE native est la conservation de l'activité enzymatique et des interactions protéine-protéine natives pendant la séparation.
-
Le SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) est la méthode la plus couramment employée pour séparer les protéines dénaturées en se basant uniquement sur le poids moléculaire. Le détergent anionique SDS se lie uniformément le long du squelette polypeptidique, conférant une charge négative constante proportionnelle à la longueur de la protéine. Dans des conditions de dénaturation (généralement par chauffage dans 0,1 % de SDS), les protéines sont linéarisées et adoptent une forme de tige allongée, ce qui permet une migration en fonction de la taille à travers la matrice de polyacrylamide. Les petites protéines traversent le gel plus rapidement que leurs homologues plus grandes. En outre, le SDS-PAGE bénéficie des principes de l'isotachophorèse, concentrant les échantillons de protéines en bandes étroites et définies et améliorant ainsi la résolution (concentration ≥ dix fois supérieure observée dans des environnements de laboratoire contrôlés).
Il est essentiel de prêter attention à la préparation de l'échantillon, à la composition du tampon et aux caractéristiques du gel pour obtenir une séparation et une reproductibilité optimales. Les composants des tampons d'échantillons typiques et leurs fonctions respectives sont résumés dans le tableau 1.
Tableau 1. Tampons de préparation d'échantillons couramment utilisés. Localisation de la protéine Tampon Objectif
| Localisation des protéines | Tampon | Rôle des composants (concentrations typiques indiquées entre parenthèses) |
| Cellule entière | Tampon non dénaturant, non ionique : tampon NP-40 : 50 mM Tris pH 8,0, 150 mM NaCl, 1 % NP-40 ou Triton X-100 |
Tris-HCl (10–50 mM) : tamponnage de la solution (contribue à la stabilité des protéines) NaCl (50–150 mM) : réduit les liaisons non spécifiques, maintient la force ionique NP-40/Triton X-100 (0,1–1 %) : détergents non ioniques, solubilisent les protéines cytosoliques et membranaires sans les dénaturer, perméabilisent les membranes et augmentent la solubilité des protéines en empêchant leur agrégation |
| Tampon dénaturant : tampon RIPA : 150 mM NaCl, 1 % NP-40 ou Triton X-100, 0,5 % désoxycholate de sodium, 0,1 % SDS, 50 mM Tris, pH 8,0 |
Désoxycholate de sodium (0,1–0,5 %) : détergent ionique, lyse les cellules et solubilise les composants cellulaires et membranaires SDS (0,1–1 %) : détergent ionique, provoque la rupture des membranes et linéarise les protéines par liaison |
|
| Cytoplasmique (soluble) |
20 mM Tris-HCl, pH 7,5 ou tampon NP-40 | Voir ci-dessus |
| Cytoplasmique (lié au cytosquelette) |
Tampon PIPES-Triton : 10 mM PIPES, 50 mM KCl, 1 % Triton X-100, 10 mM EGTA, 3 mM MgCl₂, 2 M glycérol |
PIPES (10 mM) : tamponnage de la solution KCl (50–150 mM) : réduit les liaisons non spécifiques, maintient la force ionique EGTA (1–3 mM) : inhibition des métalloprotéases dépendantes du Ca²⁺ MgCl₂ (1–5 mM) : réduit les liaisons non spécifiques ; associé au KCl, stabilise certains complexes Glycérol (1–10 %) : stabilise les protéines |
| Associé aux membranes | Tampon RIPA | Voir ci-dessus |
| Mitochondries | Tampon RIPA | Voir ci-dessus |
| Noyau | Tampon RIPA | Voir ci-dessus ; note : NP-40 et Triton X-100 ne peuvent pas lyser les membranes nucléaires |
| Localisation des protéines | Tampon |
Le pourcentage de gel requis dépend de la taille de votre protéine d'intérêt :
| Taille de la protéine | Pourcentage de gel |
| 4–40 kDa | 20% |
| 12–45 kDa | 15% |
| 10–70 kDa | 12% |
| 15–100 kDa | 10% |
| 25–100 kDa | 8% |
| 60-210 kDa | 5% |
Des gels en gradient peuvent également être utilisés :
| Taille de la protéine | Pourcentage de gel |
| 5-200 kDa | Gradient 4-12% |
| 4-200 kDa | Gradient 4-20% |
| 3.5-110 kDa | Gradient 10-20% |
Systèmes Tampons Continus versus Discontinus
Les systèmes de séparation électrophorétique peuvent être classés en deux grandes catégories : les systèmes tampons continus et les systèmes tampons discontinus (également appelés multiphasique ou isotachophoresis), chacun étant caractérisé par des configurations tampons et des dynamiques de séparation distinctes.
1. Systèmes Tampons Continus :
-
Mêmes ions tampons et même pH dans l’échantillon, le gel et les réservoirs.
-
Gel constitué d'une seule concentration d'acrylamide.
-
Les protéines se séparent directement dans le gel sans étape de concentration préalable (« stacking »).
-
Point clé : la résolution dépend fortement de petits volumes d’échantillon très concentrés.
2. Systèmes Tampons Discontinus (Multiphasique) :
-
Deux tampons différents avec pH et force ionique distincts :
-
Gel de concentration (stacking gel) : pH ~6,8, Tris–HCl faible (0,125 M), pores larges.
-
Gel de séparation (resolving gel) : pH ~8,8, Tris–HCl élevé (0,375 M), pores fins.
-
-
Tampon de migration (ex. : Tris-glycine) appliqué au-dessus du gel de concentration.
3. Mécanisme de séparation :
-
Les ions Cl⁻ agissent comme ions conducteurs (leading ions) ; les glycinate comme ions retardateurs (trailing ions).
-
Les protéines se concentrent en bandes serrées dans le gel de concentration (« effet stacking »).
-
Dans le gel de séparation, les protéines se séparent selon leur poids moléculaire en devenant les principaux porteurs de charge.
4. Avantages des Systèmes Discontinus :
-
Permet le chargement de volumes plus importants d’échantillons dilués.
-
Résolution supérieure grâce à l’effet de concentration et aux tailles de pores différenciées des gels.
-
Séparation plus efficace basée sur le poids moléculaire.
Transfert de Protéines
Après électrophorèse, les protéines sont transférées d'un gel de polyacrylamide sur une membrane. Cette étape est cruciale pour immobiliser les protéines en vue d'une détection et d'une analyse ultérieures (par exemple, par western blot).
1. Membranes Utilisées pour le Transfert de Protéines
Différentes membranes sont disponibles, chacune ayant des avantages spécifiques :
-
La nitrocellulose est la membrane la plus couramment utilisée. Elle présente une forte affinité de liaison aux protéines, peut immobiliser à la fois les protéines et les glycoprotéines, et est compatible avec les méthodes de détection chimiluminescentes, chromogéniques et fluorescentes. Cependant, elle est fragile car elle est dérivée de la cellulose traitée à l'acide nitrique.
-
Le PVDF (fluorure de polyvinylidène) offre une meilleure résistance mécanique que la nitrocellulose, ce qui le rend idéal pour le stripping et la reprobe ou pour le séquençage des protéines. Toutefois, le PVDF peut parfois entraîner une coloration de fond plus élevée, nécessitant une optimisation supplémentaire.
2. Méthodes de Transfert de Protéines
Les protéines peuvent être transférées du gel à la membrane par plusieurs méthodes :
-
Le transfert par capillarité repose sur l'action capillaire passive.
-
Le transfert par diffusion permet aux protéines de diffuser lentement vers la membrane.
-
L'électrotransfert est la méthode la plus rapide et la plus efficace.
Parmi celles-ci, l'électrotransfert est la méthode privilégiée pour sa rapidité et son efficacité.
Types d'Électrotransfert
Il existe trois types d'électrotransfert selon l'utilisation du tampon :
-
L'électrotransfert à sec utilise des composants secs sans ajout de liquide.
-
L'électrotransfert semi-sec utilise des papiers filtres imbibés de tampon de transfert entre la membrane et le gel.
-
L'électrotransfert humide immerge l'ensemble du sandwich gel-membrane dans un tampon de transfert, recommandé en particulier pour le transfert de grandes protéines.
1. Principes de l'Électrotransfert
Lors de l'électrotransfert :
-
Un champ électrique est appliqué perpendiculairement au gel.
-
Les protéines, étant chargées, migrent hors du gel vers la membrane.
-
Le gel doit être orienté vers l'électrode négative (cathode) tandis que la membrane fait face à l'électrode positive (anode).
-
Pour assurer un transfert optimal, le gel et la membrane doivent être solidement superposés, avec des papiers filtres et des tampons en fibre de chaque côté pour éviter la formation de bulles qui pourraient provoquer un transfert inégal.
a. Tampons de Transfert
Les tampons de transfert aident à maintenir la structure des protéines et favorisent leur migration pendant le transfert. Les tampons couramment utilisés sont :
-
Le tampon de Towbin (25 mM Tris, 192 mM glycine, 20 % méthanol, pH 8,3) est largement utilisé pour les transferts humides.
-
Le tampon de Bjerrum Schafer-Nielsen (48 mM Tris, 39 mM glycine, pH 9,2, 20 % méthanol) est préféré pour les transferts semi-secs.
-
Le tampon CAPS (60 mM Tris et 40 mM CAPS) est utilisé notamment pour les protéines ayant des points isoélectriques élevés.
Le méthanol dans le tampon aide à prévenir le gonflement du gel et améliore la fixation des protéines à la membrane. Toutefois, dans certains cas, des tampons sans méthanol peuvent produire des signaux de détection intenses.
Le rôle des composants du tampon de transfert est décrit dans le Tableau 2.
Table 2. Running and transfer buffer composition and role of each component
Tableau 2. Composition des tampons de migration et de transfert et rôle de chaque composant
| Composant potentiel | Rôle |
| Tampons de migration | |
| Acétate | Bonne séparation pour les protéines de masse élevée (100–500 kDa) |
| Glycine | Fournit des ions glycinate nécessaires à la formation du front traînant lors de l’électrophorèse dans le gel de concentration |
| MES | Nécessaire pour maintenir un pH relativement constant. Fournit des ions MES pour former le front traînant pendant l’électrophorèse. Le MES permet une meilleure séparation dans la gamme de basses masses moléculaires (<50 kDa). Le tampon MES-SDS migre plus rapidement que le tampon MOPS-SDS en raison du pKa plus faible du MES par rapport au MOPS. |
| MOPS | Nécessaire pour maintenir un pH relativement constant. Fournit des ions MOPS pour former le front traînant pendant l’électrophorèse. Le MOPS améliore la séparation dans les gammes de masses moyennes à élevées. La différence de mobilité ionique entre le MES et le MOPS affecte l’empilement, influençant ainsi la gamme de séparation des protéines. |
| SDS | Aide à maintenir les protéines sous une charge nette négative |
| Tricine | Nécessaire pour maintenir un pH relativement constant. Substitue la glycine dans le tampon de migration, fournissant des ions tricine pour la formation du front traînant pendant l’électrophorèse. Améliore l’empilement et la résolution des protéines de faible masse moléculaire. Idéal pour la séparation des protéines de 0,5 à 50 kDa. |
| Tris-HCl | Nécessaire pour maintenir un pH relativement constant. Fournit des ions chlorure formant le front dirigeant pendant l’électrophorèse dans le gel de concentration. Les ions hydrogène assurent la conduction électrique. |
| Tampons de transfert | |
| Tris-HCl | Nécessaire pour maintenir un pH relativement constant |
| Glycine | En absence de méthanol, aide à prévenir le gonflement du gel |
| Méthanol | Empêche le gonflement du gel pendant le transfert et améliore la fixation des protéines sur la nitrocellulose. Élimine le SDS des protéines, facilitant leur liaison aux membranes de nitrocellulose. Certains laboratoires omettent désormais le méthanol sans différence significative de résultats. Certains protocoles remplacent 20% de méthanol par 10% d’éthanol. |
| SDS | Le SDS (jusqu’à 0,1%) dans le tampon de transfert améliore l’efficacité du transfert des protéines, notamment pour les grandes protéines, mais peut réduire leur fixation à la membrane. Sur les membranes de nitrocellulose à grands pores (0,45 μm), les petites protéines dénaturées peuvent traverser la membrane. |
| CAPS | Nécessaire pour maintenir un pH relativement constant. Recommandé pour le transfert des protéines de masse élevée (>150 kDa) |
| CAPS : Acide (cyclohexylamino)-L-propane sulfonique ; MES : Acide 2-(N-morpholino)éthanesulfonique ; MOPS : Acide 3-(N-morpholino)propane sulfonique ; SDS : Dodécylsulfate de sodium ; Tris-HCl : Chlorhydrate de Tris. |
|
b. Considérations importantes
-
Les protéines de très faible poids moléculaire (<10 kDa) peuvent être transférées de manière inefficace.
-
Si le pH du tampon de transfert est inférieur au point isoélectrique de la protéine, les protéines peuvent migrer en sens inverse.
-
Des tampons spécialisés (tels que ceux à base de CAPS) sont souvent nécessaires pour transférer efficacement des protéines ayant un point isoélectrique élevé.
Étape de blocage dans le Western Blot
Après le transfert réussi des protéines sur la membrane, l'étape de blocage est essentielle pour garantir la spécificité et la clarté de la détection immunologique qui suit.

1. Objectif et importance
-
Le blocage empêche la liaison non spécifique des anticorps à la membrane, réduisant ainsi le bruit de fond et éliminant les faux positifs.
-
Il améliore la précision et la fiabilité de la détection des protéines lors du western blot.
2. Procédure de blocage
-
La membrane est incubée pendant 1 heure à température ambiante dans une solution de blocage contenant :
-
Albumine sérique bovine (BSA) ou lait écrémé en poudre, dilué dans :
-
Solution saline tamponnée au Tris avec 0,1 % de Tween 20 (TBST), ou
-
Solution saline tamponnée au phosphate avec 0,1 % de Tween 20 (PBST).
-
-
-
Les protéines présentes dans la BSA et le lait (principalement la caséine et les protéines de lactosérum) se lient aux sites inoccupés de la membrane, empêchant ainsi les interactions non spécifiques entre la membrane et les anticorps primaires ou anticorps secondaires.
3. Caractéristiques des membranes et rôle du Tween 20
-
Les membranes en nitrocellulose et en PVDF présentent de fortes affinités de liaison aux protéines principalement par interactions hydrophobes.
-
Bien que les protéines du BSA et du lait puissent s'associer faiblement aux protéines transférées, l'ajout de Tween 20 dans la solution de blocage réduit ces interactions non spécifiques, assurant un blocage plus efficace.
Incubation des anticorps et détection dans le Western Blot
1. Rôle de l'incubation des anticorps dans le Western Blot
Après le blocage de la membrane pour éviter les liaisons non spécifiques, le western blot passe à l'étape cruciale de l'incubation des anticorps. La membrane est d'abord incubée avec un anticorps primaire qui se lie spécifiquement à la protéine cible. Après avoir lavé les anticorps non liés avec des tampons tels que TBST ou PBST, un anticorps secondaire est introduit. Cet anticorps secondaire reconnaît l'anticorps primaire et est généralement conjugué à une enzyme telle que HRP (peroxydase de raifort) ou phosphatase alcaline (AP), permettant ainsi la détection du signal.
-
Anticorps primaires : Essentiels pour la liaison spécifique et sélective aux protéines cibles.
-
Anticorps secondaires : Permettent la détection et l'amplification du signal.
Astuce : Il est important d'optimiser la concentration des anticorps pour maximiser la sensibilité et réduire le bruit de fond. Notre support technique peut vous aider dans l’optimisation de vos protocoles.
Les étapes de lavage sont cruciales pour éliminer les anticorps non liés. Toutefois, un lavage excessif (>20 minutes) peut affaiblir le signal — un équilibre est donc nécessaire !
3. Caractéristiques des Membranes et Rôle du Tween 20
-
Les membranes en nitrocellulose et en PVDF présentent de fortes affinités de liaison aux protéines principalement par des interactions hydrophobes.
-
Alors que l'albumine sérique bovine (BSA) et les protéines du lait peuvent se lier faiblement aux protéines transférées, l'ajout de Tween 20 dans le tampon de blocage réduit ces interactions non spécifiques, assurant un blocage plus efficace.
2. Méthodes de Détection du Signal
Une fois la protéine cible liée par les anticorps marqués, le signal doit être détecté. Plusieurs méthodes sont disponibles :
a. Chimiluminescence (ECL)
La chimiluminescence améliorée (ECL) est la méthode de détection la plus utilisée. À l'aide d'anticorps secondaires conjugués à la HRP et de substrats ECL, de la lumière est générée et capturée soit sur film, soit à l'aide d'un système d'imagerie numérique.
-
Très grande sensibilité
-
Large gamme dynamique
-
Idéal pour détecter des protéines en faible abondance
Produits Recommandés :
Anticorps Secondaires Conjugués à la HRP
Kits de Substrats ECL
Kits de Détection ECL
b. Détection par Fluorescence
Les anticorps secondaires conjugués à des colorants fluorescents permettent une détection multiplex — ce qui signifie que vous pouvez visualiser plusieurs protéines simultanément en utilisant différents fluorophores.
-
Convient pour la détection simultanée de plusieurs cibles
-
Excellente gamme dynamique et quantification
-
Idéal lors de l'utilisation de fluorophores dans la gamme infrarouge
Produits Recommandés :
Anticorps Secondaires (Plage Visible et Infrarouge)
Systèmes d’Imagerie Fluorescente
Anticorps Fluorescents
c. Détection Chromogénique
Les méthodes chromogéniques utilisent des réactions enzymatiques pour générer un précipité coloré sur la membrane, visible à l'œil nu.
- Idéal lorsque l'équipement d'imagerie est limité.
-
Visualisation facile
-
Pas besoin d’équipement spécial
-
Idéal pour les laboratoires éducatifs et la recherche de base
Produits Recommandés :
Kits de Substrat Chromogénique (par exemple, BCIP/NBT)
d. Détection Radioactive (Moins Courante Aujourd'hui)
Les sondes radioactives étaient autrefois largement utilisées pour le western blotting, mais elles sont maintenant principalement remplacées par des méthodes plus sûres et tout aussi sensibles, comme l'ECL et la fluorescence. Le choix de la méthode de détection appropriée est essentiel pour le succès de votre Western blot et dépend de la sensibilité, de la spécificité et du type d'analyse dont vous avez besoin :
-
Sensibilité maximale ? → Choisissez la détection par Chimiluminescence Améliorée (ECL).
L'ECL offre une grande sensibilité et une large gamme dynamique, ce qui la rend idéale pour détecter des protéines en faible abondance. L'intensité du signal peut être capturée sur des films radiographiques ou des systèmes d'imagerie numériques. -
Analyse multiplex (détection de plusieurs cibles simultanément) ? → Choisissez la détection par fluorescence.
Les anticorps secondaires fluorescents permettent la détection simultanée de plusieurs protéines sans avoir besoin de retirer et de reprober les membranes. Les signaux sont hautement stables, quantitatifs, et idéaux pour les études comparatives. -
Visualisation simple et rapide ? → Choisissez la détection chromogénique.
Les systèmes chromogéniques utilisent des réactions enzyme-substrat pour produire un précipité coloré directement sur la membrane. Bien qu'ils soient moins sensibles que l'ECL ou la fluorescence, ils sont faciles à utiliser et ne nécessitent pas d'équipement d'imagerie spécialisé.
De plus, le choix des anticorps primaires et secondaires, ainsi que des substrats ou réactifs de détection, doit être compatible avec la méthode de détection choisie. Un bon couplage est essentiel pour obtenir des signaux spécifiques et forts tout en minimisant le bruit de fond.