Next-Generation Sequencing

Next-Generation Sequencing (NGS) Protocol
Introduction
Next-Generation Sequencing (NGS), also known as high-throughput sequencing, is a revolutionary technology that enables rapid and large-scale sequencing of DNA and RNA. Unlike traditional Sanger sequencing, NGS platforms process millions to billions of DNA fragments simultaneously, offering unprecedented speed, scalability, and cost-efficiency. Since its emergence in the mid-2000s, NGS has transformed genomics, enabling applications in medical research, diagnostics, agriculture, and evolutionary biology (Wang, 2021). This protocol provides a comprehensive guide for performing NGS using Illumina platforms or Ion Torrent platforms. It includes detailed steps, recommended products, and considerations for optimization. The workflow is divided into four main stages: sample extraction, library preparation, sequencing, and data analysis.
Principle of NGS
NGS involves four main steps: Sample extraction, library preparation, sequencing, and data analysis (Figure 1).
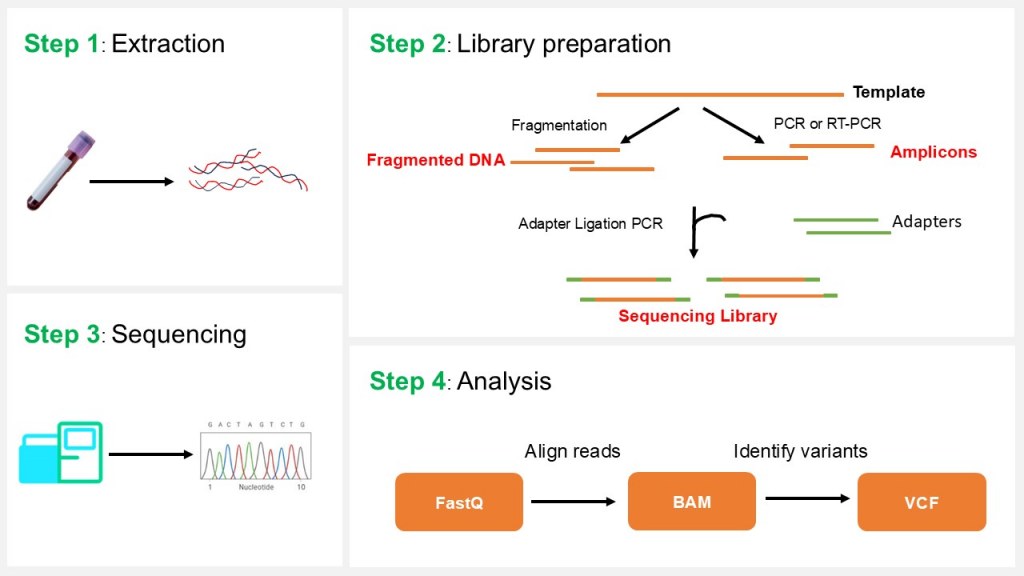
Figure 1: NGS principle
-
Sample Preparation
- DNA/RNA Extraction: High-quality nucleic acids are extracted from biological samples.
- Library Preparation: DNA or RNA is fragmented, and adapters (short, synthetic DNA sequences) are ligated to the ends of fragments to create a sequencing library. For RNA sequencing, RNA is often converted to cDNA.
- Amplification: Polymerase Chain Reaction (PCR) amplifies the library to ensure sufficient material for sequencing.
-
Sequencing
NGS platforms use various technologies, but most rely on sequencing-by-synthesis or similar methods:
- DNA fragments are immobilized on a solid surface (e.g., a flow cell).
- Fluorescently labeled nucleotides are added, and their incorporation into growing DNA strands is detected in real time.
- Common platforms include Illumina (short-read sequencing), PacBio (long-read sequencing), and Oxford Nanopore (real-time, portable sequencing).
-
Data Analysis
- Base Calling: Raw signals (e.g., fluorescence) are converted into nucleotide sequences.
- Alignment and Assembly: Reads are aligned to a reference genome or assembled de novo.
- Variant Calling and Annotation: Differences (e.g., SNPs, indels) are identified and annotated for functional impact.
- Downstream Analysis: Applications include gene expression profiling, variant discovery, or metagenomics.
NGS Protocol
Step 1: Nucleic acid isolation
1.1 Objective
Nucleic acid isolation is the critical first step in any NGS workflow, determining downstream success. High-quality DNA/RNA with purity ratios A260/280 = 1.8-2.0 and A260/230 > 2.0, (RNA Integrity Number) RIN > 8 for RNA, and concentration 10-100 ng/μL is essential to avoid PCR inhibition, adapter ligation failure, and sequencing bias. Safety: Use gloves; dispose of hazardous waste (e.g., guanidinium thiocyanate) per lab protocols.
1.2 Sample Collection and Storage
- DNA samples: Blood, tissue, cultured cells, or FFPE blocks.
- RNA samples: Fresh or frozen tissue, cultured cells, or specialized preservation buffers.
- Best practices: Avoid repeated freeze-thaw cycles. Maintain chain of custody, and assign unique sample identifiers.
1.3 DNA Isolation
DNA extraction methods vary depending on sample type and desired yield/purity. Below are detailed procedures for common sample types. Use silica-column or magnetic bead-based kits for purification. Below are detailed procedures for common sample types.
1.3.1 Blood Samples
Whole Blood / Buffy Coat
- Use EDTA-treated blood to prevent clotting.
- Lyse red blood cells (RBC) with RBC lysis buffer.
- Digest remaining cells with Proteinase K in lysis buffer (containing SDS or other detergents).
- Bind DNA to silica membrane columns or magnetic beads.
- Wash multiple times to remove salts, proteins, and contaminants.
- Elute in low-salt TE buffer or nuclease-free water.
Quality Checks
- Concentration via Qubit dsDNA HS.
- A260/A280 ~1.8.
- Optional: run 0.8% agarose gel for size verification.
1.3.2 Tissue Samples
Fresh or Frozen Tissue
- Homogenize tissue mechanically (bead mill, rotor-stator homogenizer).
- Lyse with buffer containing Proteinase K at 56°C until fully digested.
- Proceed with column or bead-based purification.
FFPE Tissue
- Deparaffinize using xylene, kit reagents, or deparaffinator.
- Reverse formalin cross-links by heating (80–90°C) with digestion buffer and Proteinase K.
- Purify using columns/beads designed for FFPE DNA.
- Expect some fragmentation due to formalin treatment; suitable for short-read NGS.
1.3.3 Cultured Cells
- Wash cells with PBS to remove medium.
- Lyse directly with Proteinase K-containing lysis buffer.
- Purify DNA with column or magnetic bead kit.
1.3.4 Tips
- Avoid excessive vortexing to prevent shearing.
- Ensure complete removal of ethanol in wash steps; residual ethanol can inhibit downstream enzymatic reactions.
1.4 RNA Isolation
RNA is highly sensitive to degradation by ubiquitous RNases. Rigorous RNase-free techniques are mandatory. For total RNA workflows, deplete rRNA using methods like poly-A selection (oligo(dT) beads) or rRNA-specific depletion to enrich mRNA (Figure 2).
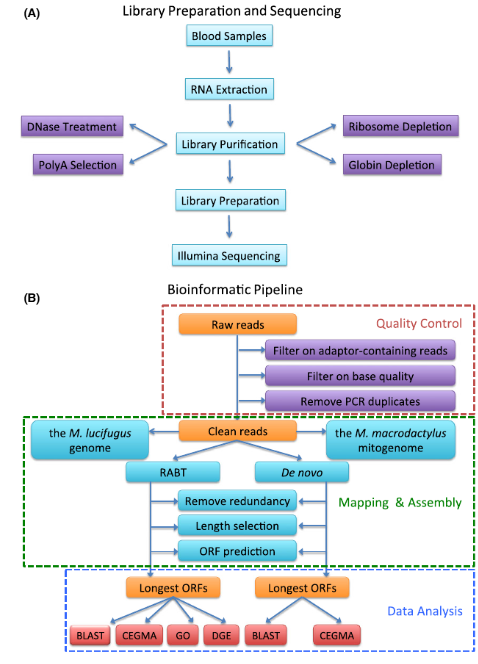
Figure 2: Workflow of RNA Extraction, Library Preparation, Sequencing, and Data Analysis. (A) Experimental steps. (B) Bioinformatic processing (Huang et al., 2016).
1.4.1 Fresh/Frozen Tissue or Cultured Cells
- Lyse in guanidinium thiocyanate-based buffer (denatures RNases).
- Optional: homogenize mechanically for tissues (bead mill, rotor-stator, or mortar/pestle with liquid nitrogen).
- Purify RNA using column or magnetic beads:
- Columns: RNA binds silica membranes under high-salt conditions.
- Magnetic beads: RNA binds to beads; wash and elute.
4. Include DNase treatment (on-column or in-solution) to remove genomic DNA.
1.4.2 Blood Samples
- Use kits designed for leukocyte RNA (e.g., PAXgene Blood RNA).
- Stabilize RNA at collection; extract promptly.
1.4.3 FFPE Samples
- Deparaffinize with xylene or kit solution.
- Reverse formalin cross-links by heating.
- Use RNA purification kits optimized for degraded RNA.
1.4.4 Quality Control
- Measure RNA concentration via Qubit RNA HS.
- Assess integrity with Bioanalyzer/TapeStation: check RIN.
- Contaminants such as phenol or salts can interfere with downstream library prep.
1.4.5 Tips
- Work quickly and keep samples on ice to reduce degradation.
- Avoid repeated freeze-thaw cycles.
- For low-input RNA, use carrier RNA if permitted by kit to improve recovery.
1.5 Troubleshooting
| Issue | Possible Cause | Solution |
| Low yield | Insufficient starting material | Increase input; optimize lysis/homogenization |
| RNA degradation | RNase contamination | Use RNase-free consumables; add RNase inhibitor; work quickly |
| High A260/230 ratio | Salt or reagent contamination | Perform additional washes; ensure ethanol fully removed |
| Fragmented DNA (unexpected) | Harsh homogenization | Reduce mechanical shearing; minimize vortexing |
| Residual gDNA in RNA prep | Incomplete DNase digestion | Increase DNase incubation; check enzyme activity |
1.6 Practical Recommendations
- Always process extraction controls to monitor contamination and consistency.
- Aliquot extracted DNA/RNA and store at −20°C or −80°C to avoid degradation.
- Document sample IDs, extraction batch, yield, purity, and integrity.
- For NGS, high-molecular-weight DNA or high-RIN RNA improves library complexity and sequencing quality.
- Avoid excessive vortexing to prevent shearing; work quickly on ice for RNA; use carrier RNA for low-input samples if compatible.
Library preparation for NGS involves converting fragmented DNA or RNA into sequencer-compatible formats by adding adapters that enable amplification, indexing, and high-throughput sequencing.
2.1. Fragmentation, End Repair, and dA-Tailing
2.1.1 Purpose
This step processes DNA to generate sequencing-ready fragments. It enzymatically fragments genomic DNA, converts heterogeneous DNA ends to blunt ends, phosphorylates 5' ends, and adds a single adenine (A) nucleotide to 3' ends (dA-tailing). The A-overhang facilitates efficient, strand-specific ligation to adapters containing thymine (T)-overhangs, crucial for Illumina platforms.
2.1.2 Reaction setup
- Combine genomic DNA with enzymatic fragmentation mix and reaction buffer in a PCR tube.
- Typical reaction volume: 20–50 µL depending on input concentration and enzyme protocol.
2.1.3 Incubation
- Fragmentation time/temperature depends on enzyme and input amount — empirically optimize.
2.1.4 Inactivation/clean-up
- After incubation, immediately proceed to purification or adapter ligation step.
- Some protocols include a heat-inactivation step at 65–70°C for 5 minutes if necessary to preserve enzyme stability during downstream steps.
2.1.5 Mechanism Details
- Fragmentation: Endonucleases introduce random double-stranded breaks yielding DNA fragments mainly in the desired size range.
- End Repair: T4 DNA polymerase fills in 5' overhangs and removes 3' overhangs to create blunt ends.
- 5' Phosphorylation: T4 polynucleotide kinase adds phosphate groups to 5' ends enabling ligation.
- dA-Tailing: DNA polymerase with terminal transferase activity adds an extra adenine nucleotide at the 3' ends of fragments, preparing them for T-overhang adapter ligation.
2.2. Adapter Ligation
Adapter ligation is a critical step in NGS library preparation where specialized adapters are enzymatically attached to both ends of fragmented DNA, enabling subsequent amplification and sequencing.
2.2.1 Purpose
Adapters facilitate attachment of DNA fragments to the sequencing flow cell and contain sequences for priming the sequencing reactions. They often include index sequences for sample multiplexing.
2.2.2 Adapter structure
Each adapter typically consists of annealed oligonucleotides shaped as a Y-adapter, with one strand binding to the flow cell and the other to the sequencing primers. Adapters have a 3' thymine (T) overhang complementary to the adenine (A) overhang added to DNA fragments during dA-tailing.
2.2.3 Reaction setup
- The DNA fragments with 3' A-overhangs are mixed with adapters in molar excess to drive efficient ligation.
- The reaction includes a DNA ligase enzyme (commonly T4 DNA ligase) and ligation buffer.
- Typical reaction volumes range from 20 to 50 µL depending on sample input.
2.2.4 Conditions
- The ligation is incubated at about 20–25°C for 15–60 minutes, depending on the protocol and enzyme used.
- A typical time is 20 minutes at 25°C for high efficiency.
2.2.5 Post-ligation
- The ligated DNA is purified to remove unincorporated adapters and enzymes, often using magnetic bead-based cleanup.
- This step prepares the ligated library for downstream amplification or sequencing.
2.3. PCR Amplification
2.3.1 Purpose
PCR amplification enriches DNA fragments that have adapters ligated to both ends, increasing the amount of sequencing-ready library. This step also introduces index sequences used for multiplexing samples in sequencing runs.
2.3.2 Reaction Setup
Combine in PCR tube:
- Adapter-ligated DNA: 10–50 ng
- Forward and reverse primers: typically 0.5 µM each
- High-fidelity DNA polymerase: 0.5–1 U
- dNTP mix: 200 µM each
- Suitable PCR buffer with MgCl2
- Bring volume to typically 50 µL with nuclease-free water
2.3.3 Thermal Cycling Conditions
- Initial denaturation: 98°C for 30 seconds
- PCR cycles (use the minimum cycles needed to obtain sufficient material (determine empirically or by qPCR), and flag PCR-free library options for high-input DNA):
- Denaturation: 98°C for 10 seconds
- Annealing: 60°C for 30 seconds
- Extension: 72°C for 30 seconds
3. Final extension: 72°C for 5 minutes
4. Hold: 4°C
Cycle number must balance amplification yield with minimal bias and errors. Over-amplification should be avoided.
2.3.4 Post-PCR Cleanup
- Purify amplified library using magnetic bead-based purification (e.g., SPRI beads) to remove primers, enzymes, and select size range (~200–600 bp).
- Validate library concentration and size distribution by Qubit fluorometry and electrophoresis or Bioanalyzer.
This PCR step is essential for generating enough material for sequencing, especially when starting with low-input DNA samples, while preserving library complexity and fidelity.
2.4. Library Purification and Size Selection
Library purification and size selection is an essential step in the NGS library preparation workflow that ensures removal of unwanted fragments, contaminants, and adapter dimers, while selecting DNA fragments of the desired size range for optimal sequencing performance.
2.4.1 Purpose
- To purify the PCR-amplified library by removing leftover primers, enzymes, salts, and adapter dimers.
- To size-select fragments within a target range (e.g., 200–600 bp) for uniform cluster generation and efficient sequencing.
2.4.2 Methods
Magnetic Bead-Based Cleanup and Size Selection (SPRI)
- Use Solid Phase Reversible Immobilization (SPRI) beads coated with silica or carboxyl groups.
- Bind DNA to beads in the presence of polyethylene glycol (PEG) and salt, which facilitates selective binding based on fragment size.
- Adjust bead-to-sample ratios to control size cutoff, enriching for desired fragment lengths.
- Mix library with appropriate volume of beads, incubate 5–10 minutes at room temperature.
- Place tubes on magnetic stand to capture beads; carefully remove supernatant containing smaller unwanted fragments.
- Wash beads twice with 70% ethanol to remove impurities.
- Air dry beads briefly (avoid over-drying).
- Elute purified DNA with low-salt buffer or nuclease-free water.
- Typically performed twice with different bead ratios for stringent size selection.
Gel Electrophoresis-Based Size Selection
- Load amplified library on agarose gel (1.5–2%) and run under appropriate voltage.
- Visualize DNA bands using safe dye.
- Excise gel slice corresponding to target fragment size (e.g., 300–500 bp).
- Extract DNA from gel slices using gel extraction kits.
- This method precisely isolates fragment size but is more labor-intensive and lower throughput.
2.4.3 Quality Control
- Quantify purified libraries using fluorometric assays (e.g., Qubit).
- Determine size distribution using Bioanalyzer, TapeStation, or gel electrophoresis.
- Confirm removal of adapter dimers and presence of target size peak before sequencing.
Step 3: Library Quality Control
3.1 Concentration
- Measure using Qubit or equivalent.
- Avoid relying solely on Nanodrop.
3.2 Size Distribution
- Analyze using Bioanalyzer, TapeStation, or Fragment Analyzer.
- Confirm expected peak and minimal adapter contamination.
Step 4: Sequencing
4.1 Library Quantification and Normalization
- Quantify purified and size-selected DNA library using fluorometric assays (e.g., Qubit) or qPCR for accurate loading.
- Normalize libraries to a uniform concentration, typically between 1-10 nM depending on sequencing platform.
4.2 Template Preparation and Cluster Generation
- Dilute the normalized library to the final concentration required by the sequencing instrument.
- Load the library onto a flow cell:
-
Illumina platforms: Amplify library fragments on flow cell surface by bridge amplification to generate dense clusters.
-
Other platforms (Ion Torrent) use beads or nanopores for template presentation.
-
-
Ensure cluster density is optimized to balance sequencing output and data quality.
4.3 Sequencing by Synthesis (Illumina Example)
- Incorporate fluorescently labeled reversible terminator nucleotides:
- Initiate sequencing cycles by addition of DNA polymerase and labeled nucleotides.
- Capture images of incorporated nucleotides after each cycle.
- Remove terminator and fluorescent labels to allow next cycle.
- Typical runs sequence 50–300 cycles depending on read length desired.
- Generate raw image files capturing cluster fluorescence across cycles.
4.4 PacBio SMRT Sequencing
- PacBio uses Single Molecule Real-Time (SMRT) sequencing, where DNA polymerase incorporates fluorescently labeled nucleotides in zero-mode waveguides. Libraries are prepared with hairpin adapters to form SMRTbell templatesCircular DNA templates used in PacBio sequencing for multiple-pass consensus reads., enabling multiple passes for high-accuracy consensus reads. Ideal for long-read applications like structural variant detection and de novo assembly.
- Template preparation: Ligate hairpin adapters to generate SMRTbell templates.
- Sequencing: Load SMRTbells onto SMRT cells; monitor fluorescence in real time.
- Applications: Resolving complex genomic regions, full-length transcript sequencing.
4.5 Oxford Nanopore Sequencing
- Oxford Nanopore sequencing detects electrical current changes as DNA/RNA passes through a protein nanopore. It supports ultra-long reads (up to 2 Mb) and real-time sequencing, suitable for field applications and metagenomics.
- Template preparation: Ligate sequencing adapters with motor proteins to guide DNA/RNA through nanopores.
- Sequencing: Load library onto flow cells; monitor current changes for base calling.
- Applications: Real-time pathogen detection, direct RNA sequencing.
4.6 Ion Torrent Sequencing
- Ion Torrent uses semiconductor-based sequencing, detecting pH changes as nucleotides are incorporated during emulsion PCR on beads. It is optimized for targeted sequencing and smaller panels.
- Template preparation: Amplify DNA on beads via emulsion PCR.
- Sequencing: Load beads onto semiconductor chips; detect pH changes.
- Applications: Cancer panel sequencing, microbial sequencing.
4.7 Platform Comparison
| Platform | Read Length | Error Rate | Throughput | Key Applications |
|---|---|---|---|---|
| Illumina | 50–300 bp | ~0.1% | High (up to 6 Tb) | WGS, RNA-seq, exome sequencing |
| PacBio | 10–20 kb | ~1–2% (raw), <0.1% (CCS) | Moderate (up to 100 Gb) | Structural variants, de novo assembly |
| Oxford Nanopore | Up to 2 Mb | ~5–10% (raw), improving with basecalling | Low to moderate (up to 50 Gb) | Real-time sequencing, metagenomics |
| Ion Torrent | 200–400 bp | ~1% | Low (up to 15 Gb) | Targeted sequencing, small panels |
4.8 Base Calling and Data Acquisition
- Convert fluorescent signals into nucleotide bases using onboard software.
- Output raw sequence reads and quality scores (FASTQ files).
4.9 Data Analysis and Quality Control
- Perform initial QC metrics: read length, quality scores, base composition, GC content.
- Align reads to reference genome or assemble de novo.
- Detect variants, structural variations, or expression levels depending on application.
Step 5: Data Processing and Bioinformatics
5.1 Primary Data Conversion
- Convert raw BCL to FASTQ (bcl2fastq, DRAGEN, or instrument software).
- Demultiplex according to index sequences.
5.2 Quality Control of Raw Reads
- FastQC: Check base quality, GC content, adapter contamination.
- MultiQC: Aggregate QC reports for multiple samples.
5.3 Read Trimming
- Remove low-quality bases and adapter sequences using Trimmomatic or Cutadapt.
5.4 Alignment
- DNA: BWA-MEM or Bowtie2 to map reads to reference genome.
- RNA: STAR or HISAT2 aligners.
- Sort and index BAM files using Samtools.
5.5 Duplicate Removal and Metrics
- Use Picard MarkDuplicates.
- Assess duplication rates, insert size distribution, and coverage metrics.
5.6 Variant Calling (for DNA)
- Use GATK Best Practices or alternatives (FreeBayes, bcftools).
- Apply quality filters and annotate variants using SnpEff or VEP.
6.7 Gene Expression Quantification (RNA-seq)
- Count reads per gene or quantify transcripts.
- Normalize and perform downstream differential expression analysis.
Quality Metrics and Troubleshooting
Critical Metrics
- Base quality Q30 ≥75%
- Mapping rate ≥90% for high-quality DNA
- Duplication rate <10–20%
- Insert size matches target range
Common Issues
- Low yield: Verify input DNA/RNA, adapter ligation efficiency, PCR efficiency.
- High adapter dimers: Perform additional bead-based cleanup.
- Low mapping rate: Check for contamination or wrong reference genome.
- High duplication: Reduce PCR cycles or increase input material.
SNPXplex: An Identity Vigilance Genotyping Kit for NGS and Genomic Analyses
The SNPXplex kit is a specialized molecular diagnostic tool designed to provide identity verification in next-generation sequencing (NGS) and genomic analyses, particularly in personalized medicine. The kit features simultaneous genotyping of 15 carefully selected single nucleotide polymorphisms (SNPs), chosen specifically to cover non-pathogenic genes to avoid population bias and ensure accurate patient identification.
Purpose and Application
SNPXplex aims to secure the integrity and traceability of patient samples in NGS workflows by confirming that the sequencing data corresponds to the correct individual. This identity vigilance is critical in clinical genomics where diagnostic precision directly affects patient management and outcomes. The test involves a straightforward PCR amplification followed by capillary electrophoresis on a sequencing platform to genotype the 15 SNP markers, enabling rapid and reliable confirmation of sample identity before clinical interpretation.
Design and SNP Selection
The 15 SNPs included in the SNPXplex panel were chosen for their neutrality with respect to disease association, ensuring that the genotyping results reflect the patient’s unique genetic fingerprint without confounding effects from pathogenic variants. This selection improves the robustness of the assay across diverse populations by minimizing population-specific allele frequency biases.
Methodology
The SNPXplex assay workflow consists of:
- DNA extraction from patient samples.
- Multiplex PCR amplification targeting the 15 SNP loci.
- Allele-specific fluorescent labeling of the PCR products.
- Capillary electrophoresis on a sequencing instrument to separate and detect allele-specific fragments.
The allele-specific fluorescent detection enables multiplex genotyping in a single run, reducing time and resource demands. The output confirms patient identity by matching the SNP profile against expected genotypes, thus ensuring sample integrity through the NGS pipeline.
Conclusion
NGS is a powerful tool, but success relies on rigorous sample handling, meticulous library preparation, careful QC at multiple stages, and precise bioinformatics workflows. Following best practices ensures high-quality data suitable for research, diagnostics, or clinical applications.
References
Wang, M., 2021. Next-generation sequencing (NGS). In Clinical molecular diagnostics (pp. 305-327).
Huang, Z., Gallot, A., Lao, N.T., Puechmaille, S.J., Foley, N.M., Jebb, D., Bekaert, M. and Teeling, E.C., 2016. A nonlethal sampling method to obtain, generate and assemble whole blood transcriptomes from small, wild mammals. Molecular Ecology Resources, 16(1), pp.150-162.