Séquençage de nouvelle génération

Protocole de séquençage de nouvelle génération (NGS)
Introduction
Le séquençage de nouvelle génération (NGS), également désigné sous le terme de séquençage à haut débit, constitue une technologie révolutionnaire qui permet le séquençage rapide et à grande échelle de l’ADN et de l’ARN. Contrairement au séquençage traditionnel de Sanger, les plateformes NGS traitent simultanément des millions à des milliards de fragments d’ADN, offrant une vitesse, une évolutivité et une rentabilité sans précédent. Depuis son apparition au milieu des années 2000, le NGS a transformé la génomique, rendant possibles des applications en recherche médicale, en diagnostic, en agriculture et en biologie évolutive (Wang, 2021). Le présent protocole fournit un guide exhaustif pour la réalisation du NGS à l’aide des plateformes Illumina ou Ion Torrent. Il inclut des étapes détaillées, des produits recommandés et des considérations relatives à l’optimisation. Le flux de travail est structuré en quatre étapes principales : extraction des échantillons, préparation des banques, séquençage et analyse des données.
Principe du NGS
Le NGS implique quatre étapes principales : extraction des échantillons, préparation des banques, séquençage et analyse des données (Figure 1).
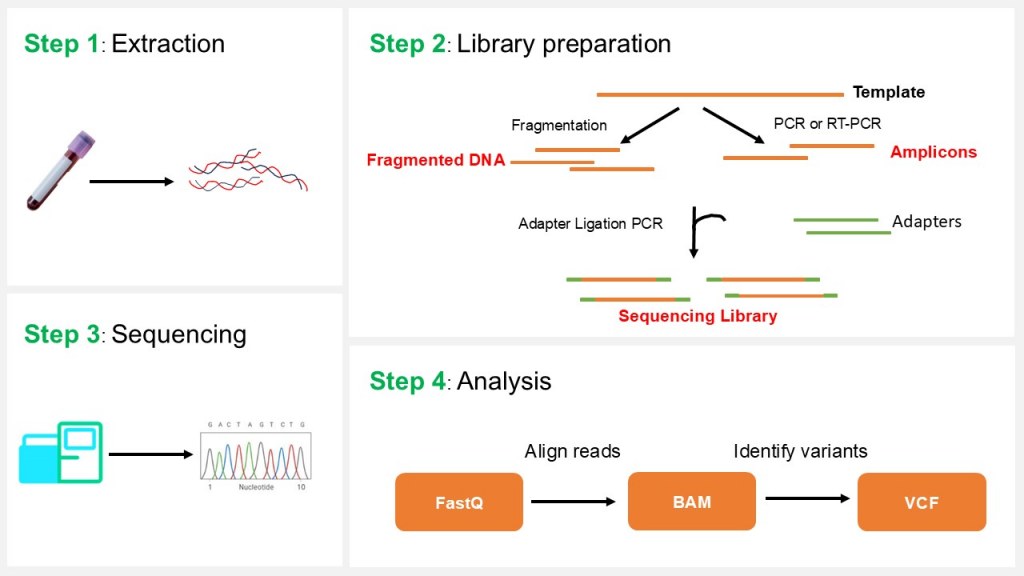
Figure 1 : Principe du NGS
-
Préparation des échantillons
- Extraction d’ADN/ARN : Des acides nucléiques de haute qualité sont extraits à partir d’échantillons biologiques.
- Préparation des banques : L’ADN ou l’ARN est fragmenté, et des adaptateurs (séquences d’ADN synthétiques courtes) sont ligaturés aux extrémités des fragments afin de constituer une banque de séquençage. Dans le cas du séquençage d’ARN, l’ARN est généralement converti en ADNc.
- Amplification : La réaction en chaîne par polymérase (PCR) amplifie la banque pour garantir une quantité suffisante de matériel pour le séquençage.
-
Séquençage
Les plateformes NGS exploitent diverses technologies, mais la plupart reposent sur le séquençage par synthèse ou des méthodes similaires :
- Les fragments d’ADN sont immobilisés sur une surface solide (par exemple, une cellule de flux).
- Des nucléotides marqués par fluorescence sont ajoutés, et leur incorporation dans les brins d’ADN en cours d’élongation est détectée en temps réel.
- Parmi les plateformes courantes figurent Illumina (séquençage à lectures courtes), PacBio (séquençage à lectures longues) et Oxford Nanopore (séquençage en temps réel, portable).
-
Analyse des données
- Appel de bases : Les signaux bruts (par exemple, fluorescence) sont convertis en séquences nucléotidiques.
- Alignement et assemblage : Les lectures sont alignées sur un génome de référence ou assemblées de novo.
- Appel de variants et annotation : Les différences (par exemple, SNP, indels) sont identifiées et annotées en fonction de leur impact fonctionnel.
- Analyse en aval : Les applications incluent le profilage de l’expression génique, la découverte de variants ou la métagénomique.
Protocole NGS
Étape 1 : Isolement des acides nucléiques
1.1 Objectif
L’isolement des acides nucléiques constitue l’étape initiale critique de tout flux de travail NGS, déterminant le succès des étapes ultérieures. Des ADN/ARN de haute qualité présentant des rapports de pureté A260/280 = 1,8-2,0 et A260/230 > 2,0, un indice d’intégrité de l’ARN (RIN) > 8 pour l’ARN, et une concentration de 10-100 ng/µL sont essentiels pour éviter l’inhibition de la PCR, l’échec de la ligature des adaptateurs et les biais de séquençage. Sécurité : Porter des gants ; éliminer les déchets dangereux (par exemple, thiocyanate de guanidinium) conformément aux protocoles de laboratoire.
1.2 Collecte et conservation des échantillons
- Échantillons d’ADN : sang, tissus, cellules cultivées ou blocs FFPE.
- Échantillons d’ARN : tissus frais ou congelés, cellules cultivées ou tampons de conservation spécialisés.
- Meilleures pratiques : éviter les cycles répétés de congélation-décongélation. Maintenir la chaîne de traçabilité et attribuer des identifiants uniques aux échantillons.
1.3 Isolement de l’ADN
Les méthodes d’extraction d’ADN varient selon le type d’échantillon et le rendement/pureté souhaités. Utiliser des kits à base de colonnes de silice ou de billes magnétiques pour la purification. Voici des procédures détaillées pour les types d’échantillons courants.
1.3.1 Échantillons de sang
Sang total / Couche leucocytaire
- Utiliser du sang traité à l’EDTA pour prévenir la coagulation.
- Lyser les globules rouges (GR) avec un tampon de lyse des GR.
- Digérer les cellules restantes avec la protéinase K dans un tampon de lyse (contenant du SDS ou d’autres détersifs).
- Fixer l’ADN sur des colonnes à membrane de silice ou des billes magnétiques.
- Effectuer plusieurs lavages pour éliminer les sels, les protéines et les contaminants.
- Élüer dans un tampon TE à faible teneur en sel ou de l’eau sans nucléase.
Contrôles de qualité
- Concentration via Qubit dsDNA HS.
- A260/A280 ≈ 1,8.
- Optionnel : électrophorèse sur gel d’agarose à 0,8 % pour vérification de la taille.
1.3.2 Échantillons de tissus
Tissu frais ou congelé
- Homogénéiser le tissu mécaniquement (broyeur à billes, homogénéisateur rotor-stator).
- Lyser avec un tampon contenant la protéinase K à 56 °C jusqu’à digestion complète.
- Poursuivre avec une purification sur colonne ou à base de billes.
Tissu FFPE
- Déparaffiner à l’aide de xylène ou de réactifs de kit.
- Inverser les réticulations formol par chauffage (80–90 °C) avec un tampon de digestion et la protéinase K.
- Purifier à l’aide de colonnes/billes conçues pour l’ADN FFPE.
- Attendre une certaine fragmentation due au traitement au formol ; adapté au NGS à lectures courtes.
1.3.3 Cellules cultivées
- Laver les cellules avec du PBS pour éliminer le milieu.
- Lyser directement avec un tampon de lyse contenant la protéinase K.
- Purifier l’ADN avec un kit à colonne ou à billes magnétiques.
1.3.4 Conseils
- Éviter un vortex excessif pour prévenir le cisaillement.
- Assurer l’élimination complète de l’éthanol lors des étapes de lavage ; l’éthanol résiduel peut inhiber les réactions enzymatiques en aval.
1.4 Isolement de l’ARN
L’ARN est hautement sensible à la dégradation par les RNases ubiquitaires. Des techniques rigoureusement exemptes de RNase sont obligatoires. Pour les flux de travail d’ARN total, appauvrir l’ARNr par des méthodes telles que la sélection poly-A (billes oligo(dT)) ou l’épuisement spécifique de l’ARNr afin d’enrichir l’ARNm (Figure 2).
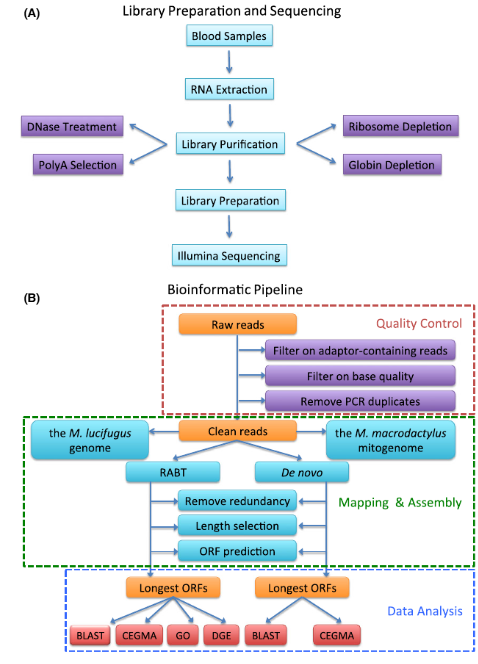
Figure 2 : Flux de travail d’extraction de l’ARN, préparation de la banque, séquençage et analyse des données. (A) Étapes expérimentales. (B) Traitement bioinformatique (Huang et al., 2016).
1.4.1 Tissu frais/congelé ou cellules cultivées
- Lyser dans un tampon à base de thiocyanate de guanidinium (dénature les RNases).
- Optionnel : homogénéiser mécaniquement les tissus (broyeur à billes, rotor-stator ou mortier/pilon avec azote liquide).
- Purifier l’ARN à l’aide de colonnes ou de billes magnétiques :
- Colonnes : l’ARN se fixe sur les membranes de silice en conditions de forte salinité.
- Billes magnétiques : l’ARN se fixe sur les billes ; laver et éluer.
4. Inclure un traitement à la DNase (sur colonne ou en solution) pour éliminer l’ADN génomique.
1.4.2 Échantillons de sang
- Utiliser des kits conçus pour l’ARN leucocytaire (par exemple, PAXgene Blood RNA).
- Stabiliser l’ARN lors de la collecte ; extraire rapidement.
1.4.3 Échantillons FFPE
- Déparaffiner avec du xylène, une solution de kit, ou deparaffinator.
- Inverser les réticulations formol par chauffage.
- Utiliser des kits de purification d’ARN optimisés pour l’ARN dégradé.
1.4.4 Contrôle de qualité
- Mesurer la concentration d’ARN via Qubit RNA HS.
- Évaluer l’intégrité avec Bioanalyzer/TapeStation : vérifier le RIN.
- Les contaminants tels que le phénol ou les sels peuvent interférer avec la préparation de la banque en aval.
1.4.5 Conseils
- Travailler rapidement et conserver les échantillons sur glace pour réduire la dégradation.
- Éviter les cycles répétés de congélation-décongélation.
- Pour un ARN à faible intrant, utiliser un ARN porteur si autorisé par le kit pour améliorer la récupération.
1.5 Dépannage
| Problème | Cause possible | Solution |
| Rendement faible | Matériau de départ insuffisant | Augmenter l’intrant ; optimiser la lyse/homogénéisation |
| Dégradation de l’ARN | Contamination par RNase | Utiliser des consommables exempts de RNase ; ajouter un inhibiteur de RNase ; travailler rapidement |
| Rapport A260/230 élevé | Contamination par sel ou réactif | Effectuer des lavages supplémentaires ; s’assurer de l’élimination complète de l’éthanol |
| ADN fragmenté (inattendu) | Homogénéisation agressive | Réduire le cisaillement mécanique ; minimiser le vortex |
| ADNg résiduel dans la préparation d’ARN | Digestion incomplète par DNase | Augmenter l’incubation à la DNase ; vérifier l’activité enzymatique |
1.6 Recommandations pratiques
- Toujours traiter des témoins d’extraction pour surveiller la contamination et la cohérence.
- Aliquoter l’ADN/ARN extrait et conserver à −20 °C ou −80 °C pour éviter la dégradation.
- Documenter les identifiants d’échantillon, le lot d’extraction, le rendement, la pureté et l’intégrité.
- Pour le NGS, un ADN de haut poids moléculaire ou un ARN à RIN élevé améliore la complexité de la banque et la qualité du séquençage.
- Éviter un vortex excessif pour prévenir le cisaillement ; travailler rapidement sur glace pour l’ARN ; utiliser un ARN porteur pour les échantillons à faible intrant si compatible.
Étape 2 : Préparation de la banque
La préparation de la banque pour le NGS consiste à convertir l’ADN ou l’ARN fragmenté en formats compatibles avec le séquenceur par l’ajout d’adaptateurs permettant l’amplification, l’indexage et le séquençage à haut débit.
2.1. Fragmentation, réparation des extrémités et ajout de dA
2.1.1 Objectif
Cette étape traite l’ADN pour générer des fragments prêts au séquençage. Elle fragmente enzymatiquement l’ADN génomique, convertit les extrémités hétérogènes en extrémités émoussées, phosphoryle les extrémités 5′ et ajoute un nucléotide adénine (A) unique aux extrémités 3′ (dA-tailing). Le surplomb A facilite une ligature efficace et spécifique au brin avec des adaptateurs à surplomb thymine (T), cruciale pour les plateformes Illumina.
2.1.2 Préparation de la réaction
- Combiner l’ADN génomique avec un mélange enzymatique de fragmentation et un tampon de réaction dans un tube PCR.
- Volume typique de réaction : 20–50 µL selon la concentration d’intrant et le protocole enzymatique.
2.1.3 Incubation
- Le temps/température de fragmentation dépend de l’enzyme et de la quantité d’intrant — optimiser empiriquement.
2.1.4 Inactivation/nettoyage
- Après incubation, passer immédiatement à la purification ou à l’étape de ligature des adaptateurs.
- Certains protocoles incluent une étape d’inactivation thermique à 65–70 °C pendant 5 minutes si nécessaire pour préserver la stabilité enzymatique lors des étapes en aval.
2.1.5 Détails mécanistiques
- Fragmentation : Endonucléases introduisent des cassures double brin aléatoires produisant des fragments d’ADN principalement dans la gamme de taille souhaitée.
- Réparation des extrémités : La polymérase T4 de l’ADN comble les surplombs 5′ et élimine les surplombs 3′ pour créer des extrémités émoussées.
- Phosphorylation 5′ : La kinase polynucléotidique T4 ajoute des groupes phosphate aux extrémités 5′ permettant la ligature.
- dA-Tailing : Une polymérase d’ADN à activité transférase terminale ajoute un nucléotide adénine supplémentaire aux extrémités 3′ des fragments, les préparant à la ligature avec des adaptateurs à surplomb T.
2.2. Ligature des adaptateurs
La ligature des adaptateurs est une étape critique de la préparation de la banque NGS au cours de laquelle des adaptateurs spécialisés sont attachés enzymatiquement aux deux extrémités de l’ADN fragmenté, permettant l’amplification et le séquençage ultérieurs.
2.2.1 Objectif
Les adaptateurs facilitent la fixation des fragments d’ADN à la cellule de flux de séquençage et contiennent des séquences pour amorcer les réactions de séquençage. Ils incluent souvent des séquences d’index pour le multiplexage des échantillons.
2.2.2 Structure des adaptateurs
Chaque adaptateur consiste typiquement en oligonucléotides appariés formant un adaptateur en Y, une branche se liant à la cellule de flux et l’autre aux amorces de séquençage. Les adaptateurs présentent un surplomb thymine (T) 3′ complémentaire du surplomb adénine (A) ajouté aux fragments d’ADN lors du dA-tailing.
2.2.3 Préparation de la réaction
- Mélanger les fragments d’ADN à surplomb A 3′ avec les adaptateurs en excès molaire pour favoriser une ligature efficace.
- La réaction inclut une ligase d’ADN (généralement la ligase T4 de l’ADN) et un tampon de ligature.
- Volumes typiques de réaction : 20 à 50 µL selon l’intrant d’échantillon.
2.2.4 Conditions
- Incuber la ligature à environ 20–25 °C pendant 15–60 minutes, selon le protocole et l’enzyme utilisée.
- Durée typique : 20 minutes à 25 °C pour une efficacité élevée.
2.2.5 Post-ligature
- Purifier l’ADN ligaturé pour éliminer les adaptateurs non incorporés et les enzymes, souvent par nettoyage à base de billes magnétiques.
- Cette étape prépare la banque ligaturée pour l’amplification ou le séquençage en aval.
2.3. Amplification PCR
2.3.1 Objectif
L’amplification PCR enrichit les fragments d’ADN auxquels des adaptateurs sont ligaturés aux deux extrémités, augmentant la quantité de banque prête au séquençage. Cette étape introduit également des séquences d’index utilisées pour le multiplexage des échantillons lors des runs de séquençage.
2.3.2 Préparation de la réaction
Combiner dans un tube PCR :
- ADN ligaturé aux adaptateurs : 10–50 ng
- Amorces directe et inverse : typiquement 0,5 µM chacune
- ADN polymérase haute fidélité : 0,5–1 U
- Mélange de dNTP : 200 µM chacun
- Tampon PCR approprié avec MgCl2
- Porter le volume à typiquement 50 µL avec de l’eau sans nucléase
2.3.3 Conditions de cyclage thermique
- Dénaturation initiale : 98 °C pendant 30 secondes
- Cycles PCR (utiliser le nombre minimal de cycles nécessaire pour obtenir suffisamment de matériel (déterminer empiriquement ou par qPCR), et envisager les options de banque sans PCR pour l’ADN à haut intrant) :
- Dénaturation : 98 °C pendant 10 secondes
- Hybridation : 60 °C pendant 30 secondes
- Élongation : 72 °C pendant 30 secondes
3. Élongation finale : 72 °C pendant 5 minutes
4. Maintien : 4 °C
Le nombre de cycles doit équilibrer le rendement d’amplification avec un biais et des erreurs minimaux. Éviter la sur-amplification.
2.3.4 Nettoyage post-PCR
- Purifier la banque amplifiée par purification à base de billes magnétiques (par exemple, billes SPRI) pour éliminer les amorces, les enzymes et sélectionner la gamme de taille (~200–600 pb).
- Valider la concentration et la distribution de taille de la banque par fluorimétrie Qubit et électrophorèse ou Bioanalyzer.
Cette étape PCR est essentielle pour générer suffisamment de matériel pour le séquençage, en particulier avec des échantillons d’ADN à faible intrant, tout en préservant la complexité et la fidélité de la banque.
2.4. Purification et sélection de taille de la banque
La purification et la sélection de taille de la banque constituent une étape essentielle du flux de travail de préparation de banque NGS qui assure l’élimination des fragments indésirables, des contaminants et des dimères d’adaptateurs, tout en sélectionnant les fragments d’ADN dans la gamme de taille souhaitée pour des performances de séquençage optimales.
2.4.1 Objectif
- Purifier la banque amplifiée par PCR en éliminant les amorces résiduelles, les enzymes, les sels et les dimères d’adaptateurs.
- Sélectionner les fragments dans une gamme cible (par exemple, 200–600 pb) pour une génération uniforme de clusters et un séquençage efficace.
2.4.2 Méthodes
Nettoyage et sélection de taille à base de billes magnétiques (SPRI)
- Utiliser des billes d’immobilisation réversible en phase solide (SPRI) recouvertes de silice ou de groupes carboxyle.
- Fixer l’ADN aux billes en présence de polyéthylène glycol (PEG) et de sel, facilitant une fixation sélective selon la taille des fragments.
- Ajuster les rapports billes/échantillon pour contrôler la coupure de taille, enrichissant les longueurs de fragments souhaitées.
- Mélanger la banque avec le volume approprié de billes, incuber 5–10 minutes à température ambiante.
- Placer les tubes sur un support magnétique pour capturer les billes ; éliminer soigneusement le surnageant contenant les petits fragments indésirables.
- Laver les billes deux fois avec 70 % d’éthanol pour éliminer les impuretés.
- Sécher brièvement les billes à l’air (éviter le sur-séchage).
- Élüer l’ADN purifié avec un tampon à faible teneur en sel ou de l’eau sans nucléase.
- Typiquement effectué deux fois avec différents rapports de billes pour une sélection de taille rigoureuse.
Sélection de taille par électrophorèse sur gel
- Charger la banque amplifiée sur un gel d’agarose (1,5–2 %) et faire migrer sous tension appropriée.
- Visualiser les bandes d’ADN à l’aide d’un colorant sûr.
- Découper la tranche de gel correspondant à la taille cible des fragments (par exemple, 300–500 pb).
- Extraire l’ADN des tranches de gel à l’aide de kits d’extraction de gel.
- Cette méthode isole précisément la taille des fragments mais est plus laborieuse et à faible débit.
2.4.3 Contrôle de qualité
- Quantifier les banques purifiées à l’aide d’essais fluorimétriques (par exemple, Qubit).
- Déterminer la distribution de taille à l’aide de Bioanalyzer, TapeStation ou électrophorèse.
- Confirmer l’élimination des dimères d’adaptateurs et la présence du pic de taille cible avant le séquençage.
Étape 3 : Contrôle de qualité de la banque
3.1 Concentration
- Mesurer à l’aide de Qubit ou équivalent.
- Éviter de se fier uniquement au Nanodrop.
3.2 Distribution de taille
- Analyser à l’aide de Bioanalyzer, TapeStation ou Fragment Analyzer.
- Confirmer le pic attendu et une contamination minimale par les adaptateurs.
Étape 4 : Séquençage
4.1 Quantification et normalisation de la banque
- Quantifier la banque d’ADN purifiée et sélectionnée en taille à l’aide d’essais fluorimétriques (par exemple, Qubit) ou de qPCR pour un chargement précis.
- Normaliser les banques à une concentration uniforme, typiquement entre 1-10 nM selon la plateforme de séquençage.
4.2 Préparation du template et génération de clusters
- Diluer la banque normalisée à la concentration finale requise par l’instrument de séquençage.
- Charger la banque sur une cellule de flux :
-
Plateformes Illumina : amplifier les fragments de banque sur la surface de la cellule de flux par amplification en pont pour générer des clusters denses.
-
Autres plateformes (Ion Torrent) utilisent des billes ou des nanopores pour la présentation du template.
-
-
S’assurer que la densité de clusters est optimisée pour équilibrer le rendement de séquençage et la qualité des données.
4.3 Séquençage par synthèse (Illumina)
- Incorporer des nucléotides terminateurs réversibles marqués par fluorescence :
- Initier les cycles de séquençage par addition de polymérase d’ADN et de nucléotides marqués.
- Capturer des images des nucléotides incorporés après chaque cycle.
- Éliminer le terminateur et les marqueurs fluorescents pour permettre le cycle suivant.
- Runs typiques : 50–300 cycles selon la longueur de lecture souhaitée.
- Générer des fichiers image bruts capturant la fluorescence des clusters à travers les cycles.
4.4 Séquençage SMRT PacBio
- PacBio utilise le séquençage en temps réel sur molécule unique (SMRT), où la polymérase d’ADN incorpore des nucléotides marqués par fluorescence dans des guides d’ondes à mode zéro. Les banques sont préparées avec des adaptateurs en épingle à cheveux pour former des templates SMRTbell, permettant plusieurs passages pour des lectures consensus de haute précision. Idéal pour les applications à lectures longues comme la détection de variants structuraux et l’assemblage de novo.
- Préparation du template : ligaturer des adaptateurs en épingle à cheveux pour générer des templates SMRTbell.
- Séquençage : charger les SMRTbell sur des cellules SMRT ; surveiller la fluorescence en temps réel.
- Applications : résolution de régions génomiques complexes, séquençage de transcrits complets.
4.5 Séquençage Oxford Nanopore
- Le séquençage Oxford Nanopore détecte les changements de courant électrique lorsque l’ADN/ARN passe à travers une nanopore protéique. Il supporte des lectures ultra-longues (jusqu’à 2 Mb) et un séquençage en temps réel, adapté aux applications sur le terrain et à la métagénomique.
- Préparation du template : ligaturer des adaptateurs de séquençage avec des protéines motrices pour guider l’ADN/ARN à travers les nanopores.
- Séquençage : charger la banque sur des cellules de flux ; surveiller les changements de courant pour l’appel de bases.
- Applications : détection en temps réel de pathogènes, séquençage direct d’ARN.
4.6 Séquençage Ion Torrent
- Ion Torrent utilise un séquençage à base de semi-conducteurs, détectant les changements de pH lors de l’incorporation de nucléotides au cours d’une PCR en émulsion sur billes. Optimisé pour le séquençage ciblé et les petits panels.
- Préparation du template : amplifier l’ADN sur billes via PCR en émulsion.
- Séquençage : charger les billes sur des puces semi-conductrices ; détecter les changements de pH.
- Applications : séquençage de panels oncologiques, séquençage microbien.
4.7 Comparaison des plateformes
| Plateforme | Longueur de lecture | Taux d’erreur | Débit | Applications clés |
|---|---|---|---|---|
| Illumina | 50–300 pb | ~0,1 % | Élevé (jusqu’à 6 Tb) | Séquençage du génome entier, RNA-seq, séquençage d’exome |
| PacBio | 10–20 kb | ~1–2 % (brut), <0,1 % (CCS) | Modéré (jusqu’à 100 Gb) | Variants structuraux, assemblage de novo |
| Oxford Nanopore | Jusqu’à 2 Mb | ~5–10 % (brut), en amélioration avec l’appel de bases | Faible à modéré (jusqu’à 50 Gb) | Séquençage en temps réel, métagénomique |
| Ion Torrent | 200–400 pb | ~1 % | Faible (jusqu’à 15 Gb) | Séquençage ciblé, petits panels |
4.8 Appel de bases et acquisition des données
- Convertir les signaux fluorescents en bases nucléotidiques à l’aide du logiciel embarqué.
- Produire des lectures de séquence brutes et des scores de qualité (fichiers FASTQ).
4.9 Analyse des données et contrôle de qualité
- Effectuer des métriques de CQ initiales : longueur de lecture, scores de qualité, composition de bases, teneur en GC.
- Aligner les lectures sur un génome de référence ou assembler de novo.
- Détecter les variants, les variations structurales ou les niveaux d’expression selon l’application.
Étape 5 : Traitement des données et bioinformatique
5.1 Conversion des données primaires
- Convertir les fichiers BCL bruts en FASTQ (bcl2fastq, DRAGEN ou logiciel de l’instrument).
- Démultiplexer selon les séquences d’index.
5.2 Contrôle qualité des lectures brutes
- FastQC : Vérifier la qualité des bases, le contenu en GC, la contamination par adaptateurs.
- MultiQC : Regrouper les rapports de contrôle qualité pour plusieurs échantillons.
5.3 Nettoyage des lectures
- Supprimer les bases de faible qualité et les séquences d’adaptateurs avec Trimmomatic ou Cutadapt.
5.4 Alignement
- ADN : BWA-MEM ou Bowtie2 pour mapper les lectures sur le génome de référence.
- ARN : Aligneurs STAR ou HISAT2.
- Trier et indexer les fichiers BAM avec Samtools.
5.5 Suppression des duplicatas et métriques
- Utiliser Picard MarkDuplicates.
- Évaluer les taux de duplication, la distribution des tailles d’insert et les métriques de couverture.
5.6 Appel de variants (pour l’ADN)
- Suivre les bonnes pratiques GATK ou alternatives (FreeBayes, bcftools).
- Appliquer des filtres de qualité et annoter les variants avec SnpEff ou VEP.
5.7 Quantification de l’expression génique (RNA-seq)
- Compter les lectures par gène ou quantifier les transcrits.
- Normaliser et effectuer une analyse d’expression différentielle en aval.
Métriques de qualité et dépannage
Métriques critiques
- Qualité de base Q30 ≥ 75 %
- Taux de mapping ≥ 90 % pour l’ADN de haute qualité
- Taux de duplication < 10–20 %
- Taille d’insert conforme à la plage cible
Problèmes courants
- Rendement faible : Vérifier l’ADN/ARN en entrée, l’efficacité de ligation des adaptateurs, l’efficacité PCR.
- Dimeres d’adaptateurs élevés : Effectuer un nettoyage supplémentaire avec billes.
- Taux de mapping faible : Vérifier la contamination ou le génome de référence incorrect.
- Duplication élevée : Réduire les cycles PCR ou augmenter la quantité de matériel en entrée.
SNPXplex : Kit de génotypage de vigilance identitaire pour le NGS et les analyses génomiques
Le kit SNPXplex est un outil de diagnostic moléculaire spécialisé conçu pour assurer la vérification d’identité dans les analyses de séquençage de nouvelle génération (NGS) et génomiques, particulièrement en médecine personnalisée. Le kit permet le génotypage simultané de 15 polymorphismes nucléotidiques simples (SNP) soigneusement sélectionnés, choisis pour cibler des gènes non pathogènes afin d’éviter les biais populationnels et garantir une identification précise du patient.
Objectif et application
SNPXplex vise à sécuriser l’intégrité et la traçabilité des échantillons de patients dans les workflows NGS en confirmant que les données de séquençage correspondent bien à l’individu concerné. Cette vigilance identitaire est cruciale en génomique clinique où la précision diagnostique impacte directement la prise en charge et les résultats des patients. Le test repose sur une simple amplification PCR suivie d’une électrophorèse capillaire sur une plateforme de séquençage pour génotyper les 15 marqueurs SNP, permettant une confirmation rapide et fiable de l’identité de l’échantillon avant l’interprétation clinique.
Conception et sélection des SNP
Les 15 SNP inclus dans le panel SNPXplex ont été choisis pour leur neutralité vis-à-vis des associations pathologiques, garantissant que les résultats de génotypage reflètent l’empreinte génétique unique du patient sans interférence de variants pathogènes. Cette sélection renforce la robustesse du test à travers des populations diverses en minimisant les biais de fréquence allélique spécifiques à certaines populations.
Méthodologie
Le workflow du test SNPXplex comprend :
- Extraction d’ADN à partir des échantillons de patients.
- Amplification PCR multiplexée ciblant les 15 loci SNP.
- Marquage fluorescent spécifique d’allèle des produits PCR.
- Électrophorèse capillaire sur un séquenceur pour séparer et détecter les fragments spécifiques d’allèle.
La détection fluorescente spécifique d’allèle permet un génotypage multiplexé en une seule réaction, réduisant le temps et les ressources nécessaires. Le résultat confirme l’identité du patient en comparant le profil SNP aux génotypes attendus, assurant ainsi l’intégrité de l’échantillon tout au long du pipeline NGS.
Conclusion
Le NGS est un outil puissant, mais son succès repose sur une manipulation rigoureuse des échantillons, une préparation minutieuse des librairies, un contrôle qualité soigneux à chaque étape et des workflows bioinformatiques précis. Le respect des bonnes pratiques garantit des données de haute qualité adaptées à la recherche, au diagnostic ou aux applications cliniques.
Références
Wang, M., 2021. Séquençage de nouvelle génération (NGS). In Diagnostic moléculaire clinique (pp. 305-327).
Huang, Z., Gallot, A., Lao, N.T., Puechmaille, S.J., Foley, N.M., Jebb, D., Bekaert, M. et Teeling, E.C., 2016. Une méthode d’échantillonnage non létal pour obtenir, générer et assembler des transcriptomes sanguins complets à partir de petits mammifères sauvages. Molecular Ecology Resources, 16(1), pp.150-162.